










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Endocrinología
versión On-line ISSN 1561-2953
Rev Cubana Endocrinol vol.29 no.2 Ciudad de la Habana mayo.-ago. 2018
PRESENTACIÓN DE CASO
Síndrome de Cowden
Cowden syndrome
Adalberto Luis Infante Amorós, Wladimir Mauricio Vélez Ruiz, Ana del Carmen Argüelles Zayas, Reinaldo Denis de Armas
Hospital Clinicoquirúrgico "Hermanos Ameijeiras". La Habana, Cuba.
RESUMEN
El síndrome de Cowden es una enfermedad hereditaria, de transmisión autosómica dominante, caracterizada por la presencia de múltiples hamartomas y nódulos en la piel y la mucosa oral, junto con anomalías en mamas, tiroides y pólipos en el tracto gastrointestinal, con un riesgo incrementado de tumores malignos. Se reporta un caso de una paciente con diagnóstico de bocio tóxico nodular, y que presentaba -por los antecedentes y estudios realizados- manifestaciones compatibles con el síndrome de Cowden. El síndrome de Cowden es el síndrome hamartomatoso tumoral del phosphatase and tensinhomolog mejor descrito hasta el momento. Los pacientes con él tienen lesiones mucocutáneas características y un elevado riesgo de cáncer de mama, tiroides, endometrio, colorrectal y renal, así como varias manifestaciones benignas como macrocefalia y gangliocitomadisplásico del cerebelo. Es importante el diagnóstico precoz de este síndrome y el seguimiento a largo plazo, dado el alto riesgo de desarrollar tumores malignos.
Palabras clave: síndrome de Cowden; hipertiroidismo; hamartomas; pólipos de colon.
ABSTRACT
Cowden syndrome is a hereditary disease, of autosomal dominant transmission, and characterized by the presence of multiple hamartomas and nodules in the skin and oral mucosa, and also with abnormalities in the breast, thyroid, and polyps in the gastrointestinal tract with an increased risk of malignant tumors. It is reported a case of a patient with a diagnosis of toxic nodular goiter, and who presented -due to the antecedents and studies carried out- manifestations compatible with the Cowden syndrome. Cowden syndrome is the hamartomatous tumor syndrome of phosphatase and tensin homolog which is better described so far. Patients having it present characteristic mucocutaneous lesions and a high risk of breast, thyroid, endometrial, colorectal and renal cancers, as well as several benign manifestations such as macrocephaly and gangliocytoma of the cerebellum. Early diagnosis of this syndrome and long-term follow-up are important given the high risk of developing malignant tumors.
Keywords: Cowden syndrome; hyperthyroidism; hamartomas; colon polyps.
INTRODUCCIÓN
El síndrome de Cowden (SC), también llamado síndrome de hamartomas múltiples o síndrome neoplásico, es una enfermedad hereditaria, de transmisión autosómica dominante con penetrancia incompleta y expresividad variable. Fue descrita por primera vez por Lloyd y Dennis en 1963, que la denominaron con el nombre de su paciente. Forma parte del síndrome hamartomatoso tumoral del phosphatase and tensinhomolog (PTEN), que además está comprendido por el síndrome de Bannayan-Riley-Ruvalcaba, los cuales son desórdenes que están asociados a mutaciones en el gen supresor tumoral PTEN y un riesgo incrementado de tumores benignos y malignos.1
A pesar de que los datos son limitados, la prevalencia estimada del SC es de 1 caso por cada 200 000 a 250 000 personas.2,3
El gen del PTEN regula negativamente la enzima fosfatidil-inositol-3-quinasa-AKT, la cual es de gran importancia para la proliferación celular, progresión del ciclo celular y la apoptosis. La pérdida de la función de este gen contribuye a la oncogénesis, y por tal razón, el PTEN, el cual se encuentra localizado en el cromosoma 10q23, y es considerado un gen supresor tumoral.4
El SC es el síndrome hamartomatoso tumoral del PTEN mejor descrito hasta el momento. Los pacientes con este síndrome tienen lesiones mucocutáneas características y un elevado riesgo de cáncer de mama, tiroides, endometrio, colorrectal y renal, así como varias manifestaciones benignas como macrocefalia y gangliocitomadisplásico del cerebelo.5,6
El caso clínico que se presenta a continuación se trata de una paciente que presentó hipertiroidismo, asociado además a manifestaciones tumorales renales e intestinales.
PRESENTACIÓN DEL CASO
Paciente femenina, de 59 años de edad, mestiza, de procedencia urbana, con antecedentes patológicos personales de hipertensión arterial esencial, diabetes mellitus tipo 2, y de carcinoma endometrial, para lo cual se le realizó histerectomía en el año 1995; así como, antecedentes familiares de dos hermanas con tumor renal de etiología no precisada, y abuelo paterno con carcinoma de pulmón.
La paciente acudió inicialmente a la consulta de Endocrinología por presentar pérdida de peso de aproximadamente 20 kg en 6 meses, sin relación con disminución de ingesta alimentaria, acompañado de ansiedad, palpitaciones, disnea al esfuerzo, temblor involuntario de ambas manos, y diarreas líquidas en número de 3 a 4 al día, alternando con periodos de constipación.
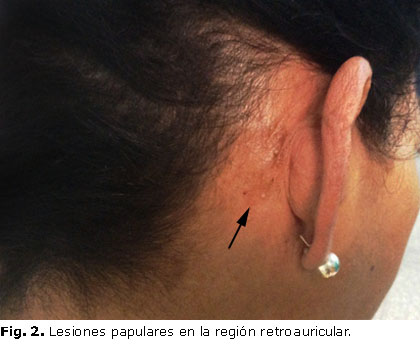
En la exploración física se constató: lesiones papulares en cara, región retroauricular y anterior del cuello (figuras 1, 2 y 3), piel húmeda y caliente, tremor, bocio grado 2 (visible con el cuello en posición normal y palpable) de consistencia elástica, superficie nodular en el lóbulo izquierdo, circunferencia cefálica de 54 cm y frecuencia cardiaca de 114 latidos por minuto.


Se indicaron estudios complementarios que informaron: niveles elevados de triyodotironina (T3) y tetrayodotironina o tiroxina (T4), supresión de la hormona estimulante del tiroides (TSH), presencia de anticuerpos antitiroideos antiperoxidasa (anti-TPO) ligeramente positivos (tabla 1). Además, se realizó captación de yodo que resultó compatible con hipertiroidismo.

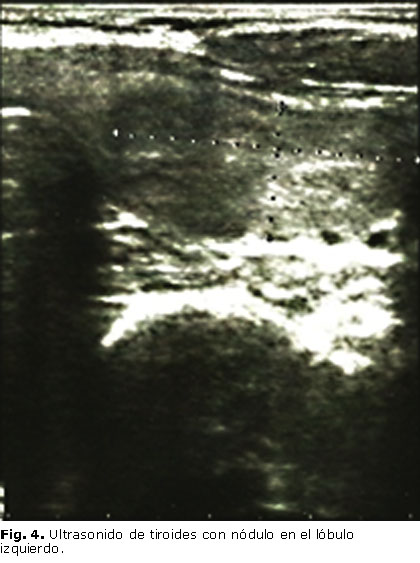
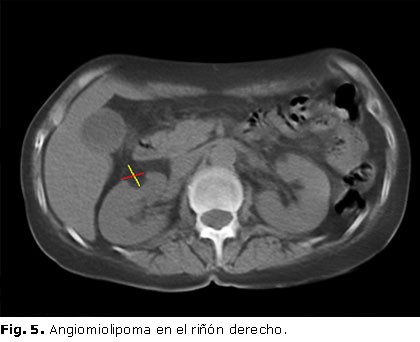
En el ultrasonido de la región anterior del cuello se describió tiroides heterogénea, con un ligero aumento de tamaño y presencia de nódulo hipoecogénico que ocupa todo el lóbulo izquierdo, que mide 30 x 12 mm (figura 4); mientras que, en estudio ultrasonográfico abdominal, se destacó la presencia de nódulo sólido de 21 x 15 mm hacia la cara anterior del riñón derecho compatible con hamartoma, que fue corroborado mediante tomografía axial computarizada (TAC) de abdomen, que informó presencia de imagen nodular de densidad grasa, que mide 14 x 10 mm, a nivel del polo superior del riñón derecho, sugestiva de angiomiolipoma (figura 5).
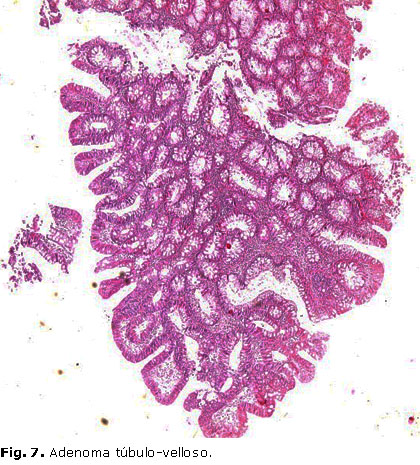
Con los diagnósticos antes mencionados, y ante la sospecha de SC, se decidieron realizar estudios complementarios adicionales. El ultrasonido de mama resultó normal; la colonoscopia informó presencia de lesión plana del colon transverso, múltiples pólipos de colon transverso y recto, diverticulosis de colon izquierdo, a los cuales se les realizó biopsia que resultó en adenoma tubular (figura 6) y adenoma túbulo-velloso de 0,4 cm (figura 7). Se solicitó además TAC de cráneo, que detalló aracnoidocele sellar, y biopsia de la lesión de piel informó quiste folicular (milium).

La paciente fue diagnosticada con un adenoma tóxico, angiomiolipoma y pólipos de colon. Se indicó tratamiento con propiltiouracilo a una dosis de 100 mg cada 8 horas (300 mg al día) y propranolol 40 mg cada 8 horas.
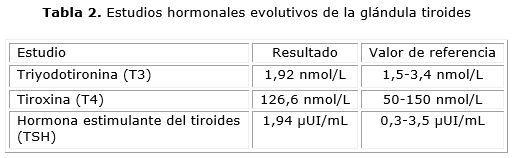
A los 3 meses posteriores al inicio del tratamiento con antitiroideos de síntesis y beta bloqueadores, la paciente acudió a consulta evolutiva, en la cual se evidenció mejoría clínica, dada por remisión de todos los síntomas y signos asociados a tirotoxicosis, así como disminución de los niveles séricos de hormonas tiroideas y normalización de los niveles de TSH (tabla 2).
DISCUSIÓN
El SC presenta un ligero predominio en el sexo femenino (60 %), y afecta preferentemente a la raza blanca (96 %).6 El caso clínico coincide con dichas características. En cuanto a la edad, suele manifestarse antes de la tercera década de la vida.6,7
Los criterios diagnósticos para el SC fueron propuestos en 1998 por el Consorcio Internacional de Cowden, y están divididos en: criterios patognomónicos, criterios mayores y criterios menores (tabla 3).7
Para establecer el diagnóstico de SC deben cumplir uno de los criterios operacionales siguientes:
1. Lesiones mucocutáneas patognomónicas, solo si hay:
- Seis o más pápulas faciales, de las cuales 3 o más deben ser triquilemomas, o
- pápulas faciales cutáneas y papilomatosis de la mucosa oral, o
- papilomatosis de la mucosa oral y queratosis acral, o
- seis o más queratosis palmo-plantares
2. Dos criterios mayores, de los cuales uno debe ser macrocefalia o enfermedad de Lhermitte-Duclos
3. Un criterio mayor y 3 menores
4. Cuatro criterios menores
A pesar de que la paciente no presenta las lesiones mucocutáneas patognomónicas corroboradas por biopsia, sí cumple con el criterio diagnóstico operacional 3, en el que el criterio mayor lo constituye el antecedente personal de carcinoma endometrial, y los criterios menores son: enfermedad tiroidea, pólipos intestinales y angiomiolipoma (tumor renal).
Las pruebas para identificar la mutación del PTEN, que incluyen el análisis de la secuencia de una región codificante entera y el análisis de la deleción/duplicación, están disponibles en muchos laboratorios clínicos y de genética molecular.8
El SC tiene efectos en casi todos los órganos y sistemas, por lo cual es necesario que el tratamiento sea realizado por un equipo multidisciplinario, que además debe incluir consejería genética.9,10
Dado el alto riesgo de malignidad, la vigilancia del cáncer es el punto principal del manejo médico. La consejería y educación genética con respecto al síndrome y las posibles manifestaciones, incluyen las recomendaciones siguientes:11
- Examen físico detallado anual, con atención particular en mama y tiroides, iniciando a los 18 años de edad, o 5 años después de la edad más joven de diagnóstico de un componente de cáncer en la familia.
- Ultrasonido de tiroides a los 18 años de edad, y considerar repetirlo anualmente a partir de ese momento.
- Colonoscopia a partir de los 35 años de edad, y luego cada 5 a 10 años, o más frecuentemente si aparecen síntomas de pólipos.
- Examen dermatológico anual.
La presentación de este caso clínico tiene gran importancia, dado que además de ser infrecuente, el valor diagnóstico de la identificación precoz de lesiones tumorales a nivel de diferentes órganos, permitiría canalizar la conducta diagnóstica y terapéutica de manera más oportuna y multidisciplinaria, logrando de esta manera disminuir la morbilidad y mortalidad por cáncer en pacientes en los cuales el componente de malignidad tumoral está dado por factores genéticos.
Conflicto de intereses
Los autores declaran no tener conflicto de intereses en la realización del estudio.
REFERENCIAS BIBLIOGRÁFICAS
1. Elia A, Amato C, Bottitta M. An atypical patien with Cowden syndrome and PTEN gene mutation presenting with cortical malformation and focal epilepsy. Brains & Development. 2012;34:873.
2. Nelen MR, Kremer H, Konings IB. Novel PTEN mutations in patients with Cowden disease: absence of cleargenotype-phenotype correlations. Eur J Hum Genet. 1999;7:267.
3. Jelovac D, Park BH. PTEN promoter silencing and Cowden syndrome: The role of epigenetic regulation of KILLIN. JAMA. 2010 Dec 22/29;304:2744.
4. Keniry M, Parsons R. The role of PTEN signalling perturbations in cancer and in targeted therapy. Oncogene. 2008;27:5477.
5. Bennett KL, Mester J, Eng C. Germline epigenetic regulation of KILLIN in Cowden and Cowden-like syndrome. JAMA. 2010;304:2724.
6. Almenar R, Bagán J, Sebastián, Milián M. Síndrome de Cowden: presentación de un caso clínico con lesiones orales. Anales de Medicina Interna. 2001;18:8.
7. Kenneth Y, Bennett KL. Cowden and Cowden-like Syndrome: Tales from the other strand. JAMA. 2010;22:29.
8. Tan MH, Mester J, Peterson C. A clinical scoring system for selection of patients for PTEN mutation testing is proposed on the basis of a prospective study of 3 042 probands. Am J Hum Genet. 2011;88:42.
9. Pilarski R, Stephens JA, Noss R. Predicting PTEN mutations: an evaluation of Cowden syndrome and Bannayan-Riley-Ruvalcaba syndrome clinical features. J Med Genet. 2011;48:505.
10. McBride KL, Varga EA, Pastore MT. Confirmation study of PTEN mutations among individuals with autism or developmental delays/mental retardation and macrocephaly. Autism Res. 2010;3:137.
11. Ni Y, He X, Chen J. Germline SDHx variants modify breast and thyroid cancer risks in Cowden and Cowden-like syndrome via FAD/NAD-dependant destabilization of p53. Hum Mol Genet. 2012;21:300.
Recibido: 22 de noviembre de 2017.
Aprobado: 14 de marzo de 2018.


{kind=link}