










 Servicios personalizados
Servicios personalizados
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
Introducción
El síndrome de Mc Cune-Albright (SMA) es una entidad clínica poco frecuente. En su descripción original se asociaba con la displasia fibrosa poliostótica (DFP), con manchas de color café con leche y también con la pubertad precoz;1 posteriormente se han reconocido otros estados de hiperfunción hormonal como el hipertiroidismo la hiperprolactinemia y la hipersomatotropinemia.1,2 En la actualidad, a la presencia de la tríada original se le conoce como la forma clásica del SMA, mientras que la forma no clásica incluye la afección ósea asociada, las manchas de color café con leche y/o la hiperfunción hormonal.1
Aunque por lo general se diagnostica en la infancia, la edad y forma de presentación varían de manera considerable. Lo anterior es particularmente cierto para los síndromes endocrinos asociados pues un paciente puede o no presentar pubertad precoz en la infancia y en el transcurso de su vida desarrollar otra hiperfunción hormonal.
La patogénesis molecular del SMA es muy compleja. La alteración básica ocurre en el gen GNAS1, el cual presenta mutaciones somáticas activadoras (llamadas también ganadoras de función) que se adquieren de manera poscigótica y que explican las alteraciones óseas, cutáneas y endocrinas del síndrome.2,3
Por su naturaleza sistémica, el manejo del SMA requiere de un equipo multidisciplinario de especialistas que incluye pediatras, internistas, endocrinólogos, neurocirujanos, ortopédicos, entre otros.
El objetivo de esta investigación fue describir las características clínicas de una paciente con síndrome de Mc Cune-Albright con una pubertad precoz.
Presentación del caso
Paciente de 7 años, femenina, raza negra, resultado de un 4to embarazo con antecedentes prenatales, natales y posnatales negativos.
A los 3 años de edad presentó sangrado vulvar en forma de manchas rojas, de 3 días de duración, negando la mamá traumatismos de ningún tipo. Al examen físico se constató máculas hipercrómicas amplias que ocupaban hemicuerpo derecho, mamas Tanner II, peso: 17 Kg, talla: 88 cm, (Talla/edad >97 percentil, que se corresponde con una edad de 5.4 años.
Se realizaron complementarios hormonales FSH: 0,2 UI/L, LH: no detectable, Estradiol: 3265pg/ml// 1863pg/ml encontrándose muy elevado.
Estudios de imagen: US ginecológico: aumento del volumen ovárico, US abdominal: sin alteraciones, Radiografía de edad ósea: acelerada, y estudio citogenético. Se diagnostica síndrome de Mc Cune Albright expresado por pubertad precoz, manchas café con leche y lesiones óseas. Con todos los elementos antes mencionados, se decidió frenar el desarrollo puberal. De inicio se administró Arimidex (Anastrozol) 1 mg/ día x 2 semanas y luego Tamoxifeno 20 mg/día con el cual se ha mantenido hasta la fecha. (2014-2018) y apoyo psicológico para la familia y la paciente.
Fue vista en reconsulta a la edad de 7.3 años, al examen físico se encontró:
Fascie: Asimetría fascial, frente prominente, macrocráneo.
Piel: Máculas hipercrómicas en hemicuerpo derecho que no pasan la línea media, algunas máculas hipocrómicas en el mismo hemicuerpo en abdomen y miembro inferior.
Mucosas: Normocoloreadas y húmedas
FR: 20 x min. Murmullo vesicular normal. No se auscultan estertores.
FC: 91x min. Ruidos cardíacos rítmicos y bien golpeados. No se auscultan soplos. Buen llene capilar. Pulsos periféricos presentes.
TA: 106/60 mmhg.
Abdomen: Blando, depresible, no doloroso a la palpación, no se palpa visceromegalia. Ruidos hidroaéreos presentes.
SOMA: Deformidad en valgus en ambos miembros inferiores
SNC: Bien orientada, no signos de focalización neurológica. No signos meníngeos.
Investigaciones realizadas
Hemograma con diferencial:
Hb: 119 g/l Hto: 0.38
Leucocitos: 5.3 x 109/L
Polimorfonucleares: 38 % Linfocitos: 57 %
Monocitos: 2 %
Eosinófilos: 3 %
Eritrosedimentación: 30 mm/h
Proteína C reactiva cuantitativa: 276,6 mg/L
Glucemia: 5.0 mmol/L
Creatinina: 40 umol/L
Urea: 5.8 mmol/L
Ca: 2.06 mmol/L
Fósforo: 0.45 mmol/L
TGO: 15 U/L
TGP: 31 U/L
Fosfatasa alcalina: 1506 u/l
Gasometría: PH: 7.36 pCO2: 44.9 mmhg pO2: 30.4 mmhg EB:0.1 CHCO3: 23.1 mmol/l
Ionograma: K: 4.5 mEq/L
Na:142 mEq/L
Ca:1.24 mEq/L
Cl: 109 mEq/L
TSH: 1.62 mUI/L T4: 134.2 nmol/L
FSH: 1.8 UI/L
LH: 0.27 UI/L
Testosterona: 1.21 nmol/L
Cortisol inicial: 575.0 nmol/L
Se repite cortisol basal: 247.9 nmol/L
11:00 pm: 106 nmol/L
Prolactina: 237.1 mU/L
Calcio en orina: 0.44 mmol/ 24 h
Fósforo en orina: 12 mmol/24h
Imagenológicos
Radiografía de edad ósea: Entre 7 y 8 años (normal)
Survey óseo:
Pelvis: Incurvación en varo del eje longitudinal de ambos fémures, con áreas de aspecto de vidrio esmerilado en cavidad medular.
Cúbito y rodilla: Áreas de esclerosis en porción medular bilateral.
Mano izquierda: expansión de la diáfisis con áreas de rarefacción y afinamiento cortical.
Cráneo: Engrosamiento del Diploe con elevada esclerosis en región frontal que alcanza regiones orbitarias y malares.
Ultrasonido abdominal y ginecológico: Normal.
RMN de Cráneo: Normal (fig).
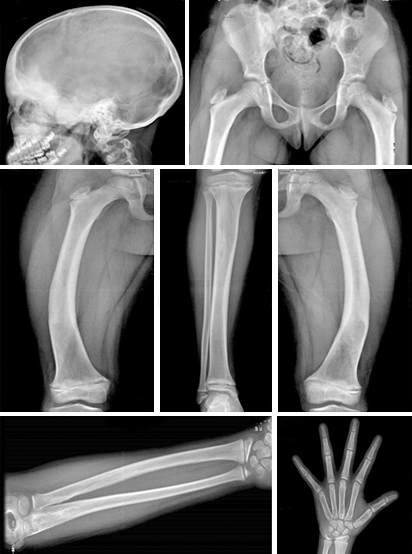
Interconsultas realizadas
Estomatología: Se diagnostica retraso del brote dentario y movilidad del 51 y 61. Se indica radiografía panorámica y se decidió exodoncia de estos.
Máxilo facial: La asimetría facial propia de la enfermedad de base no necesita corrección quirúrgica, se propuso evaluación al finalizar la pubertad.
Ortopedia: La deformidad en varo que presenta es propia de la enfermedad de base. En el momento de evaluación no tenía criterio de tratamiento correctivo quirúrgico, se propuso nueva evaluación al final de la pubertad.
Oftalmología: En la evaluación inicial hubo sospecha de glaucoma, por lo observado en el fondo de ojo, excavación 0.6 en OD y 0.1-0.2 en OI con presiones oculares iniciales OD: 21mmhg y OI: 20 mmhg, con poca cooperación. Se sugirió chequeo periódico con frecuencia trimestral y se indicaron cristales por defecto refractivo.
Genética: Por tratarse de una enfermedad por mosaisismo, están descritas mutaciones puntuales, que no han sido exploradas en esta paciente o que no existen evidencias documentadas de haber sido exploradas o buscadas. Se sugirió estudio genético para confirmación.
Otorrinolaringología: Se realizó evaluación de la niña por referir disminución de la audición. Se comprobó al examen físico tapón de cerumen, se procedió a su extracción, logrando recuperación de la audición.
Endocrinología: Se realizó evaluación clínica completa y estudio hormonal y en equipo multidisciplinario se decidió mantener tratamiento con Tamoxifeno hasta la edad del comienzo fisiológico de la pubertad (8 años) ya que la respuesta clínica había sido favorable, no hubo progresión del estadio mamario, no se incrementó el vello sexual, no reapareció el sangrado menstrual, no se aceleró la maduración esquelética. Al momento de la reevaluación estaba eutiroidea, los valores de cortisol fueron normales, la prolactina normal, el ritmo de crecimiento adecuado (talla por debajo de la media, pero en percentil normal), sólo signos de pubertad precoz periférica estáticos.
Tratamiento indicado: Tamoxifeno 20 mg diario hasta los 8 años.
Discusión
El SMA fue descrito en forma separada por los grupos de Mc Cune y Albright en la década de 1930.1,5 Esta entidad se debe a mutaciones activadoras del gen GNAS1, que se encuentra en el cromosoma 20q13.2-13.3 y codifica la proteína Gsα.2,6 Dicha proteína es una subunidad de las proteínas G con función estimuladora, que se expresa en forma ubicua y cuya función es activar la adenilato ciclasa para generar AMP cíclico (AMPC).2,7 El GNAS1 presenta una impronta genómica compleja que genera diferentes productos génicos a partir de los alelos paterno y materno, a través de la utilización de promotores alternativos improntados en forma opuesta.8 La impronta del gen GNAS1 es tejido específica. En la hipófisis, la expresión de GNAS1 depende por completo del alelo materno; mientras que en la tiroides y en las gónadas, alrededor del 20-30 % de la transcripción de este gen es contribuida por el alelo paterno.9,10,11
Las mutaciones de GNAS1 en el SMA ocurren a nivel somático y poscigótico, por lo que no existe transmisión vertical y sus manifestaciones son distintas en los diferentes tejidos, dependiendo del momento y el tipo de célula en que se producen las mutaciones, es decir, los pacientes son mosaicos somáticos.12 Es probable que las mutaciones fueran letales si se encontraran en la línea germinal y, al parecer, una presentación más severa del síndrome se relaciona con la aparición más temprana de la mutación en la etapa poscigótica.8
Las mutaciones encontradas en este síndrome inhiben la actividad guanosina trifosfatasa de Gsα, por lo que las células afectadas tienen actividad constitutiva de adenilato ciclasa y, por tanto, producción irrestricta de AMPc.2,7,8,10 Debido al fenómeno de impronta genómica, se podría especular que las manifestaciones del síndrome deberían ser diferentes de acuerdo con el origen del alelo de GNAS1 que contiene la mutación.
Hasta el momento, solo se ha demostrado que la acromegalia ocurre exclusivamente en pacientes con mutaciones en el alelo materno, mientras que para el resto de los componentes del síndrome no se ha identificado una relación entre el alelo mutado y el patrón de alteraciones clínicas.9,10,11
Este síndrome tiene una prevalencia aproximada de entre 1/100 000 y 1/1 000 000 en la población general, mientras que la displasia fibrosa ósea, en especial en su variante monostótica, es mucho más vista.2,8 Es más frecuente en mujeres que en hombres, aunque se desconoce el motivo de esta diferencia de género.4 La pubertad precoz y la displasia fibrosa ósea son los datos de presentación más frecuentes y las causas más comunes por las que los pacientes solicitan atención médica. Sin embargo, en realidad son más comunes las manchas color café con leche, aunque en ocasiones, suelen pasar desapercibidas.3
En una serie de 113 pacientes (91 % mujeres) en los que se realizó la detección de mutaciones en GNAS1 se describió la presentación clásica de pubertad precoz, manchas café con leche y displasia fibrosa poliostótica en solo 24 % de los pacientes al momento del diagnóstico. En un tercio de ellos mostró dos de los tres componentes, siendo el más común, la presencia de manchas café con leche. El resto de los pacientes mostró sólo un componente del síndrome, generalmente pubertad precoz.7
En la displasia fibrosa ósea asociada al SMA, la base del cráneo y los huesos largos son los sitios más frecuentemente involucrados. La afectación suele ser unilateral y generalmente afecta varios huesos (forma poliostótica). Algunos pacientes desarrollan lesiones en un solo hueso (forma monostótica) y, muy raras veces, afección de todo el esqueleto (forma panostótica). Esta afección consiste en lesiones focales compuestas principalmente por células estromales inmaduras de la médula ósea, espículas de tejido óseo inmaduro, osteoide no mineralizado y, en ocasiones, islas de cartílago hialino que se expanden desde la cavidad medular hacia el hueso cortical circundante, con osteoclastos en su periferia.12 Dichas células estromales inician su diferenciación hacia la línea osteogénica, pero en lugar de diferenciarse completamente, proliferan al parecer bajo el efecto continuo de la hormona paratiroidea, dando lugar a las lesiones.13
Las manifestaciones iniciales son dolor, claudicación de una extremidad o una fractura patológica.13 Los estudios radiográficos muestran lesiones de aspecto quístico o lítico, que son de radiopacidad heterogénea en niños menores de dos años. En niños mayores, son clásicas las lesiones en «vidrio esmerilado», mientras que en adultos aparece esclerosis en los bordes de las lesiones. La extensión es de la médula a la corteza ósea, generalmente se respetan las epífisis y puede existir deformación de los huesos que sostienen peso, siendo clásica la deformación del fémur en «bastón de pastor».
A nivel cráneo-facial aparecen protuberancias o asimetría facial y, cuando las lesiones se expanden, pueden causar disfunción de nervios craneales, con afección visual o auditiva, lo que ocurre principalmente en pacientes con sobreproducción de hormona de crecimiento en forma concomitante.
La displasia fibrosa ósea comienza en etapas tempranas de la vida y para los 15 años generalmente se ha establecido 90 % de la extensión total de la enfermedad, y la aparición de las lesiones nuevas es muy poco frecuente en la edad adulta. Las regiones óseas más frecuentemente involucradas son fémur proximal, y la base del cráneo, aunque cualquier región puede estar afectada. La incidencia de fracturas patológicas es mayor entre los 5 y 10 años de edad.14
A nivel de huesos largos, se recomienda el tratamiento quirúrgico con colocación de prótesis intramedulares, preferentemente a edades tempranas, con el fin de disminuir la probabilidad de fracturas y la deformidad de las estructuras óseas. El tratamiento quirúrgico de las lesiones cráneo-faciales está indicado cuando hay compromiso de nervios craneales, dolor intenso y desfiguración severa.
El tratamiento con bifosfonatos, principalmente el pamidronato, es útil para aliviar el dolor; sin embargo, tiene escaso efecto sobre la progresión de la enfermedad. La evolución de las lesiones puede ser modificada por otros componentes del síndrome como la pubertad precoz (talla baja), la hipersomatotropinemia (aumento del tamaño de las lesiones) y la hiperfosfaturia (aumento del riesgo de fracturas). La transformación maligna de las lesiones óseas es muy rara aunque puede ocurrir en relación con la exposición a radiaciones ionizantes.15
Las manchas café con leche se deben a una proliferación activa de los melanocitos y a la consecuente hiperproducción de melanina. No son exclusivas de este síndrome, también existen en forma aislada y asociadas con otros síndromes genéticos. Son lesiones benignas, sin potencial aparente de progresión a malignidad. Estas manchas de color café con leche que se asocian con el SMA son de número y morfología variables. Generalmente de gran diámetro, casi siempre tienen bordes irregulares (en «costa de Maine») y se hacen más evidentes con la edad y con la exposición a la luz solar. Están limitadas a un hemicuerpo, que corresponde al involucro óseo, no suelen atravesar la línea media y se presentan principalmente en la parte posterior del cuello, el tórax y los glúteos. Se distribuyen siguiendo las líneas de Blaschko, que representan el patrón de migración de las células del ectodermo durante la etapa embrionaria.16
La pubertad precoz es la manifestación endocrina más común en pacientes con SMA 4 y es más común en las niñas que en los niños. Consiste en hiperfunción gonadal autónoma (pubertad precoz periférica), aunque puede desarrollarse posteriormente hiperfunción hipofisaria (pubertad precoz central), que disminuye la respuesta al tratamiento inicialmente instituido. Aunque la pubertad precoz en estos pacientes suele presentarse con evolución intermitente y largos periodos de remisión, algunos desarrollan progresión rápida, con aceleración del crecimiento y la maduración esquelética, que comprometen la talla final.17
Si bien, la medroxiprogesterona y el ketoconazol pueden inhibir los ciclos menstruales, no está demostrado que tengan efecto sobre la maduración ósea. Los inhibidores de la aromatasa se han considerado como una estrategia terapéutica útil en la pubertad precoz periférica; sin embargo, un estudio reciente que incluyó a 28 niñas reportó que el tratamiento con anastrozol durante un año fue inefectivo para el control de esta alteración endocrina en el contexto del síndrome de Mc Cune-Albright.18
Por otro lado, en una serie de 25 niñas con pubertad precoz y síndrome de Mc Cune-Albright, el tratamiento con tamoxifeno a 12 meses demostró una disminución de los episodios de hemorragia menstrual, así como buen control de la velocidad de crecimiento y la maduración ósea, aunque este tratamiento no tuvo efecto sobre el volumen ovárico y el tamaño uterino.17
En los pacientes que desarrollan activación del gonadostato, el tratamiento de la pubertad precoz periférica debe combinarse con análogos de la hormona liberadora de gonadotrofinas de acción prolongada. En las niñas con respuesta inadecuada al tratamiento, la resección quirúrgica de los folículos ováricos hiperfuncionantes puede ayudar a controlar la enfermedad. Sin embargo, puede existir recidiva después de la cirugía, por lo que esta estrategia se reserva para los pacientes que no responden al tratamiento farmacológico.
El hipertiroidismo es común, se encuentra en 30 y 50 % de los casos, aunque algunos pacientes presentan enfermedad subclínica. Independientemente de la presencia de hipertiroidismo clínico, en pacientes con SMA existe elevación en la relación T3-T4, debida a un aumento de la actividad de las desyodinasas D1 y D2 mediada por la producción constitutiva de adenosín monofosfato cíclico.19 El tratamiento con tionamidas puede ser eficaz, pero en estos pacientes prácticamente nunca se produce remisión espontánea de la hiperfunción tiroidea, por lo que se debe instaurar alguna forma de tratamiento definitivo (cirugía o yodo radiactivo).3
La producción excesiva de hormona de crecimiento se encuentra en alrededor de 20 % de las pacientes con SMA, aunque solo algunos de ellos presentan un adenoma hipofisario detectable por estudios de imagen. Este es uno de los cuatro síndromes genéticos que son causantes de acromegalia (los otros tres son: la neoplasia endocrina múltiple tipo 1, el complejo de Carney y la acromegalia familiar). En la hipófisis se puede encontrar hiperplasia de lactotropos y somatotropos, de manera similar a lo que ocurre en el complejo de Carney. El exceso de GH puede empeorar las lesiones cráneo-faciales, con disminución de la agudeza visual y auditiva. Además de que ha sido implicado en la transformación sarcomatosa de las lesiones por displasia fibrosa ósea.20 En un estudio de 12 pacientes con sobreproducción de hormona del crecimiento y SMA se detectó hiperprolactinemia en 92 % de los casos y adenoma hipofisario en 33 % de ellos. Se describió compromiso visual o auditivo en 33 % de estos pacientes, en comparación con 4 % en pacientes con el síndrome, pero sin exceso de GH.21
El tratamiento quirúrgico es poco factible debido a la alteración de las estructuras óseas que dificulta el abordaje, así como, a la presencia de múltiples zonas de hiperplasia diseminadas en la hipófisis en la mayoría de los pacientes, en lugar de un adenoma bien localizado, por lo que se requiere hipofisectomía.
Existe buena respuesta al tratamiento con cabergolina y octreótide por separado o combinados, aunque generalmente se logra control parcial de la enfermedad. Se ha propuesto el tratamiento con antagonistas del receptor de hormona del crecimiento como una medida que posiblemente sea más eficaz.20
En una serie de cinco pacientes con exceso de GH y SMA resistentes a dosis máximas de análogos de la somatostatina, se obtuvo control de la enfermedad en la totalidad de los casos tras el tratamiento combinado con radioterapia y pegvisomant.22
El síndrome de Cushing en pacientes con síndrome de McCune-Albright es muy poco frecuente (7 %). Se debe a la activación constitutiva del receptor de ACTH en la corteza suprarrenal, y si bien la presencia de hipercortisolismo impone mayor riesgo cardiovascular y metabólico en estos pacientes, se han descrito casos de resolución espontánea.23
El raquitismo u osteomalacia con hipofosfatemia e hiperfosfaturia se asocian con el SMA y debe buscarse en forma intencionada esta alteración, ya que, de pasar desapercibida, puede empeorar el pronóstico de las lesiones óseas. Existen dos mecanismos causales propuestos que no se excluyen entre sí: el aumento en la producción de AMPc en el túbulo contorneado proximal renal, con disminución de la reabsorción de fósforo 2 y la producción del factor de crecimiento de fibroblastos-23 (FGF-23), un conocido factor fosfatúrico, por las lesiones de displasia fibrosa ósea.
El tratamiento consiste en la administración de dosis altas de calcitriol y fosfato vía oral.8 Algunos pacientes con síndrome de Mc Cune-Albright presentan hiperparatiroidismo con hiperplasia de las glándulas paratiroides. No se ha demostrado que la hiperfunción se deba a las mutaciones en GNAS1, sino que se trata de un hiperparatiroidismo secundario debido a deficiencia de vitamina D, que puede empeorar las lesiones óseas.
Otras manifestaciones clínicas no endocrinas e inusuales de este síndrome son hiperplasia del timo, pólipos gastrointestinales,3 alteraciones hepatobiliares, y cardiovasculares. Cuando existe cardiopatía, esta se presenta con hipertrofia, taquicardia persistente e incluso muerte súbita,23 el síndrome Mc Cune Albright debe sospecharse en neonatos o lactantes con colestasis sin causa aparente.7 Los pacientes que presentan estas anomalías casi siempre tienen displasia fibrosa ósea extensa y con cierta frecuencia hipercortisolismo, por lo que aumenta su morbilidad y mortalidad. Se ha encontrado asociación entre el síndrome Mc Cune-Albright y un incremento del riesgo para desarrollar cáncer de tiroides y de mama.13
El diagnóstico diferencial debe realizarse con las siguientes afecciones:24
Displasia fibrosa monostótica
Neurofibromatosis
Hipertiroidismo
Síndrome de Cushing
Terapia con glucocorticoides
Terapia con hormonas tiroideas
Gigantismo y acromegalia
Raquitismo hipofosfatémico
Pubertad precoz de tipo central
Tumor funcionante de ovario
Síndrome de Mazabraud
Enfermedad de Paget
Síndrome de Proteus
Entre los estudios complementarios de laboratorio clínico deben incluirse:25,26
Calcio y fósforo séricos, Fosfatasa alcalina sérica
N-telopéptidos e hidroxiprolina urinarios
Osteocalcina sérica
Enzimas hepáticas
Hormonas tiroideas y TSH
ACTH sérica y Cortisol urinario
Hormona de crecimiento
Estradiol sérico, FSH y LH, testosterona sérica, en caso de pubertad precoz.
Los estudios imagenológicos deben incluir:12
Ultrasonidos de la pelvis, para detectar y medir quistes de ovarios (generalmente grandes y unilaterales) así como de las partes blandas, para detectar tumoraciones.
Tomografía axial computarizada de abdomen y de cráneo, para detectar hiperplasia suprarrenal y atrapamiento de nervios intracraneales.
Survey óseo radiográfico y determinación de la edad ósea.
Survey óseo gammagráfico, para detectar sitios asintomáticos.
Se revisó y presentó el caso de una paciente de 7 años de edad con pubertad precoz periférica. En estudios imagenológicos se aprecian deformidades óseas compatibles con displasia poliostótica y en el contexto clínico de presentación se realiza en diagnóstico de síndrome de Mc Cune Albright (SMA). La displasia fibrosa poliostótica no es una entidad frecuente en el grupo etario expuesto, su importancia radica en la presentación de una alteración benigna del tejido óseo, de baja prevalencia caracterizada por la sustitución del hueso por tejido óseo fibroso. Su gran espectro clínico y genético hace pensar que dicha condición sea explicada por más de una alteración genética.
La presentación del síndrome de Mc Cune Albright es un ejemplo de la heterogeneidad genética de esta enfermedad y la diversidad de síntomas óseos, cutáneos y endocrinos que pueden identificarse. Aunque esta entidad es de carácter benigno y la prevalencia es extremadamente baja, el inicio puberal precoz y el compromiso de la talla final pueden producir impacto psicológico en la calidad de vida y en el desarrollo normal del individuo. Estos elementos, por tanto, exigen la detección temprana de la enfermedad, siendo un ejemplo de la necesidad del manejo multidisciplinario para el tratamiento de los síntomas desde su diagnóstico.

