










 Servicios personalizados
Servicios personalizados Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
El síndrome de ovario poliquístico (SOP) es el ejemplo típico de un trastorno multifactorial complejo. En su génesis intervienen múltiples factores sistémicos y locales que tienen una relación multidireccional. La presencia de unos favorece la expresión de otros y viceversa, generando una secuencia de eventos cuya temporalidad resulta difícil de precisar. Por ello, a pesar de haberse realizado numerosos estudios, muchas cuestiones permanecen aún sin dilucidar y persiste cierta confusión e incertidumbre.1,2,3,4
El entendimiento de que se trata de un síndrome y no de una enfermedad es crucial para comprender la gran variabilidad en los mecanismos que se relacionan con su origen y desarrollo o la complejidad de cómo se estructura la interrelación entre ellos. El síndrome incluye etiologías diversas, que se presentan con intensidad variable y se asocian a condiciones desencadenantes o agravantes, también diversas, lo que determina que sea muy heterogéneo.8
Se realizó una revisión bibliográfica tipo estado del arte para describir el enfoque actual sobre las causas y los mecanismos involucrados en el origen y desarrollo del síndrome de ovario poliquístico.
Métodos
Se revisaron alrededor de 250 artículos que se obtuvieron de las bases PubMed, Medline, Scielo y Google Académico. Se emplearon como motores de búsqueda las frases “síndrome de ovario poliquístico”, “fisiopatología y síndrome de ovario poliquístico”, “etiopatogenia y síndrome de ovario poliquístico”, tanto en español como en inglés, o se identificaron como recurrentes en las fuentes consultadas. Se seleccionaron aquellos que a criterio de la autora aportaban información más valiosa, sobre todo, los publicados en los últimos cinco años.
Etiopatogenia del síndrome de ovario poliquístico
La etiología exacta del SOP no se conoce completamente. No obstante, la evidencia disponible sugiere que intervienen factores genéticos y ambientales, de cuya interacción derivan alteraciones endocrinas que condicionan la expresión fenotípica del síndrome.5,6,7,8
Factores genéticos
La hipótesis sobre el origen genético del SOP surge de la observación de mayor prevalencia del síndrome, o sus componentes en miembros de una familia o en gemelas homocigóticas, así como de evidencias que apoyan la teoría del origen prenatal del SOP. A partir de ello, diversos estudios han confirmado que se presenta con un patrón de agregación familiar de alta heredabilidad.8,9,10,11
Se han identificado múltiples polimorfismos genéticos de susceptibilidad para diversos elementos del síndrome, los que es común que se asocien.12,13) Se han descrito cambios en la expresión de genes implicados en la esteroidogénesis, la foliculogénesis, la calidad ovocitaria, el control del eje hipotálamo-hipófisis-ovario, la receptividad endometrial, el receptor de FSH (FSHR), el de andrógenos (AR), el de la hormona antimulleriana (AMH), la resistencia a la insulina (RI), y el estado pro inflamatorio, entre otros.14,15,16) En los últimos años se señala, además alteraciones de genes mitocondriales que indican nuevas vías patogénicas.17,18
El modelo de herencia no está definido, por un tiempo se postuló la transmisión autosómica dominante con trasmisión predominantemente paterna. Sin embargo, se ha observado heterogeneidad fenotípica en una misma familia y para muchos genes candidatos no se ha podido demostrar su reproducibilidad o su relación con mayor susceptibilidad para el SOP.19,20) Asimismo, se han descrito alteraciones epigenéticas que persisten en la línea celular germinal, con transmisión transgeneracional.17,21,22) Por ello, la mayoría de los autores concuerdan en que el SOP es un rasgo genético complejo altamente heredable, cuya etiología es poligénica con expresividad variable, con fuerte influencia de factores ambientales, étnicos o geográficos; con una amplia gama de variantes genéticas.6,7,8,12
Factores ambientales
La presencia de éstos desempeña un papel decisivo en la etiología del SOP. En mujeres con susceptibilidad genética, la exposición pre o posnatal a diversas condiciones favorece o amplifica la expresión de alteraciones fisiopatológicas intrínsecas o genera daño epigenético. Actúan entonces como factores de riesgo, desencadenantes o agravantes.4,5,6) Entre las exposiciones prenatales reconocidas sobresalen la malnutrición o daño fetal y la exposición intrauterina a hiperandrogenismo.5,6,22) De las exposiciones posnatales, la obesidad se considera la más relevante porque puede agravar prácticamente todos los mecanismos fisiopatogénicos del SOP y porque muchos de los restantes factores ambientales median su acción a través de ella.4,5,6
El bajo peso al nacer y la prematurez se mencionan como factores de riesgo para desarrollar SOP desde las primeras descripciones del síndrome, aunque algunos estudios prospectivos no han podido probar esta relación.23,24) Se postula que la exposición a un ambiente nutricional intrauterino insuficiente provoca cambios epigenéticos que condicionan un fenotipo “ahorrador” que las hace más susceptibles de padecer RI tras el nacimiento y los trastornos metabólicos y reproductivos asociados a ésta, incluyendo el SOP.11,23) Asimismo, en modelos animales y en humanos se ha descrito que la exposición prenatal temprana a exceso de andrógenos (de origen materno o exógeno), por influencia ambiental o mecanismos epigenéticos, puede inducir una “programación fetal” que condiciona cambios permanentes en la fisiología reproductiva y resulta en fenotipos similares al SOP.3,24,25) En hijas de mujeres con hiperandrogenismo en el embarazo es mayor la frecuencia de alteraciones endocrinas o metabólicas y SOP.11,26
En relación con la obesidad se conoce que tiene una relación bidireccional con la RI y el estado proinflamatorio se asocia a mayor concentración de andrógenos circulantes y alteración en la secreción de gonadotropinas, lo que le suma vías fisiopatogénicas a la disfunción endocrina y metabólica intrínseca del síndrome.27,28) En mujeres con SOP se han reportado alteraciones específicas del tejido adiposo como: mayor adiposidad abdominal independiente del peso corporal, producción anormal de adipocitocinas, alteración de la lipólisis estimulada por catecolaminas, el metabolismo de los ácidos grasos libres y la función del transportador intracelular de glucosa 4 (GLUT4), entre otras.8
Las dietas ricas en carbohidratos y grasas, el sedentarismo y los estilos de vida no saludables, no se relacionan de forma directa con la etiología del SOP, pero se consideran factores coadyuvantes porque conducen a obesidad o a descontrol metabólico. La importancia de focalizar la atención en estos radica en que son factores modificables, lo que permite acciones preventivas.29
Entre los factores emergentes se encuentran la exposición a disruptores endocrinos como químicos industriales (Bisfenol A y similares), fármacos (dietiletilbestrol, valproato de magnesio, insulina), pesticidas organofosforados, fitoestrógenos, contaminantes ambientales en contextos con gran polución y la deficiencia de vitamina D.7,30,31
Factores endocrinos
De las alteraciones endocrinas presentes en el SOP sobresalen tres, muy interrelacionadas entre sí: 1) el trastorno en la secreción de gonadotropinas y la regulación neuroendocrina del eje hipotálamo-hipófisis-ovario, 2) la disfunción de la esteroidogénesis y foliculogénesis ovárica, y 3) la resistencia a la insulina, con la consecuente hiperinsulinemia.5,6,7,8,10
A nivel hipotálamo-hipofisario, se ha demostrado aumento en la frecuencia y amplitud de los pulsos de secreción de la hormona liberadora de gonadotropinas (GnRH), en la secreción de hormona luteinizante (LH) y, en ocasiones, niveles circulantes elevados de esta última. La hormona estimulante del folículo (FSH) suele ser normal o tener una disminución relativa que se evidencia en aumento de la relación LH/FSH. Existe resistencia ala retroalimentación negativa hipotalámica de las hormonas sexuales. Las células de la teca responden de forma exagerada a la LH y las de la granulosa tienen una respuesta aberrante a FSH (hiperrespuesta en folículos preantrales y resistencia en estadios posteriores).5,8,9,32) Se discute si el SOP se asocia a hiperprolactinemia.33
Concerniente a las alteraciones de la esteroidogénesis, existe evidencia de disfunción enzimática ovárica y suprarrenal. Se manifiesta por incremento en la actividad de la enzima citocromo P450c17, que cataliza la acción de las enzimas 17 alfa hidroxilasa y 17-20 liasa e induce mayor producción de andrógenos. La síntesis de estradiol puede ser normal o disminuida (por menor actividad de las aromatasas) y se pierden las variaciones cíclicas normales, lo que lleva a un estado de “estro continuo”, con niveles que corresponden a los de fase folicular temprana a media. La estrona aumenta, por aromatización del exceso de androstenediona. Todo ello incrementa la relación andrógenos/estrógenos y estrona/estradiol. La anovulación se asocia a progesterona disminuida.10,34,35,36
Los folículos muestran alteraciones cuantitativas y cualitativas. Las mujeres con SOP nacen con mayor número de folículos y en edad reproductiva tienen 2-6 veces más folículos en desarrollo que en un ovario normal. Predominan (a diferencia de otras causas de folículos múltiples) los preantrales o antral pequeño. La granulosa exhibe cambios degenerativos y grados variables de luteinización y la teca es hiperplásica. El desarrollo folicular anormal es de las características distintivas del SOP. Existe mayor reclutamiento folicular, pero la selección del folículo dominante y la atresia se inhiben, lo que determina que se acumule un exceso de folículos activos.10,37
En condiciones normales todo folículo que deja de crecer en estado preantral sufre atresia. En el SOP no ocurre así, quedan detenidos en su desarrollo. Este fenómeno conocido como “arresto folicular”, es único del SOP. Los folículos detienen su crecimiento en estadio antral medio, no progresan, pero no son atrésicos ni muestran signos de apoptosis, son viables y esteroidogénicamente muy activos. Las células de la granulosa disminuyen en número, pero son funcionalmente robustas. La membrana basal se mantiene íntegra y no ocurre colapso folicular. El acúmulo progresivo de fluido folicular expande el antro que queda claramente delimitado por una capa lineal de la granulosa y la teca engrosada, lo que le confiere apariencia quística, de donde deriva la denominación del síndrome.38,39
El disturbio en la foliculogénesis incluye además alteraciones en la calidad ovocitaria, comprobadas en modelos animales y en mujeres a las que se les realiza reproducción asistida.5,6,7,8) Al comparar ovocitos de mujeres con SOP y de mujeres con función ovárica normal, de buena calidad y similar morfología, se han demostrado patrones de expresión global de genes diferentes,40 defectos en la meiosis y alteración de la ultraestructura y función mitocondrial del ovocito, así como deficiencias en la señal bidireccional célula de la granulosa-ovocito.41,42
Del mismo modo, se han descrito alteraciones en múltiples factores locales con influencia en el crecimiento folicular, la selección del folículo dominante, la esteroidogénesis y la calidad de los ovocitos. Entre ellos ha cobrado importancia creciente en los últimos años la AMH, que se encuentra aumentada en las mujeres con SOP desde el nacimiento, es proporcional al número de folículos preantrales y guarda relación con los niveles de andrógenos y LH. Por su función fisiológica en el control del desarrollo de los folículos primarios y el reclutamiento, podría explicar algunos de los mecanismos hasta ahora no bien precisados en la génesis de las alteraciones de la foliculogénesis propias del SOP.43,44,45
También se ha reportado aumento de la inhibina B, la proteína ósea morfogénica 15 (BMP-15), la relación folistatina/activina, los factores de crecimiento similares a la insulina (IGF), de crecimiento epidérmico (EGF), de necrosis tumoral α (TNF-α), de crecimiento de neurotropina (NGF) e interleucinas (IL). También figuran la disminución del factor de crecimiento y diferenciación 9 (GDF-9), de crecimiento endotelial vascular (VEGF) y la proteína transportadora de IGF (IGF-BP). Además, se señala incremento del estrés oxidativo y la acumulación de productos finales de la glucosilación avanzada (AGE) en las células de la granulosa y la teca.5,7,36,46
La RI y la hiperinsulinemia compensatoria están presentes en cerca de la mitad de las pacientes con SOP, independientemente del peso corporal. Se manifiestan desde la niñez o pubertad temprana y preceden al hiperandrogenismo bioquímico. Su etiología exacta sigue sin esclarecerse, pero se acepta que se trata de un trastorno en la señal post receptor temprana. Se han identificado alteraciones genéticas y epigenéticas relacionadas con ésta y puede exacerbarse por la obesidad y otros factores ambientales.6,7,47,48,49
Fisiopatología
La comprensión de los mecanismos que llevan al desarrollo del SOP permanece fragmentaria y no totalmente dilucidada. Se ha intentado atribuir el rol principal a: la disfunción hipotálamo-hipofisaria, el defecto en la esteroidogénesis (ovárica o adrenal) y/o la foliculogénesis, o la RI. No existe un hallazgo constante, por lo que se han objetado todas las hipótesis formuladas. Se propone que, como otros elementos del síndrome, la fisiopatología es también heterogénea y no hay un mecanismo único, sino varias vías que conducen a un fenotipo común.9,49,50
Suma complejidad al entendimiento que, por ser el SOP un trastorno multigénico y multifactorial pueden presentarse diversas variantes genéticas y asociarse a disímiles factores ambientales que con independencia de cuál sea el evento primario interactúan, se superponen y se agravan mutuamente, por lo que resulta muy difícil determinar qué es causa y qué es consecuencia.
Disfunción en la esteroidogénesis y la foliculogénesis
Existe cada vez más evidencia que apunta a que las anomalías cardinales del SOP son principalmente ováricas. En animales y en transexuales se ha demostrado que la administración de andrógenos induce la morfología ovárica característica del síndrome,51,52) estudios moleculares evidencian la disfunción de las células foliculares53) y la práctica clínica señala al hiperandrogenismo como el hallazgo más constante y el atributo más relevante del SOP. La mayoría de los autores concuerdan en que el hiperandrogenismo ovárico funcional (HOF) es la alteración fundamental en las variantes típicas del SOP, como un fenómeno primario o como consecuencia de otros factores etiológicos.37,43,54,55
La secuencia de eventos fisiopatogénicos del HOF presumiblemente se inicia en la hiperactividad intrínseca de las enzimas esteroidogénicas, que lleva a aumento en la producción de andrógenos en el ovario. El hiperandrogenismo intraovárico, por un mecanismo paracrino y autocrino, condiciona la foliculogénesis alterada y el incremento del pool de folículos activos, lo que provoca alteración de factores intraováricos e hiperandrogenemia. El exceso de andrógenos circulantes, por un mecanismo endocrino, altera los pulsos de secreción de GnRH e induce aumento selectivo de LH, que estimula la teca, incrementa más la síntesis de andrógenos y crea un círculo vicioso. La relación positiva bidireccional entre hiperinsulinemia e hiperandrogenismo pudiera agravar lo anterior.5,36,49,55
El papel del aumento de los andrógenos suprarrenales en el desarrollo del SOP no está claro, pero se pudiera explicar por los efectos de la hiperandrogenemia a nivel ovárico e hipotálamo-hipofisario, que induce disfunción en la foliculogénesis y las consecuencias que de ello derivan.5,8,50
Disfunción hipotálamo-hipofisaria
La hipótesis que señala la disfunción neuroendocrina como el elemento primario en la fisiopatogénesis del SOP se desestima por la inconstancia del incremento de LH en estas mujeres, lo que dificulta que se le pueda atribuir un rol principal. Sin embargo, se acepta que puede ser la vía en algunos casos.
La ruta fisiopatogénica se origina en el aumento de la frecuencia y amplitud de los pulsos secretorios de GnRH y el subsiguiente incremento de LH. Se formulan como posibles responsables de estas alteraciones el aumento de la sensibilidad hipofisaria a la GnRH, el estímulo de la secreción mantenida de estrógenos sobre la síntesis de gonadotropinas o receptores de GnRH, la hiperinsulinemia, el aumento de la fracción libre de los IGF o la disociación en las vías dopaminérgicas y opioidérgicas hipotalámicos (intrínseca o secundaria a otros factores).5,15,50,56
La LH, principal regulador de la síntesis de andrógenos, estimula las células de la teca, donde induce la expresión de genes que codifican la actividad de las enzimas esteroidogénicas, lo que incrementa la síntesis de andrógenos y provoca trastornos en la foliculogénesis por las vías ya descritas. La hiperandrogenemia resultante agrava la disfunción neuroendocrina y crea un círculo vicioso.8,32,36
Resistencia a la insulina
Se considera que la RI pudiera ser la alteración primaria solo en casos específicos, debido a que no es un hallazgo constante en las mujeres con SOP. No todas las mujeres con RI desarrollan SOP y existen estudios que sugieren que las anomalías intrínsecas en los ovarios y/o la secreción de gonadotropinas son un prerrequisito para que se manifiesten las acciones reproductivas de la insulina.47) No obstante, se reconoce que cuando está presente agrava el SOP y tiene un rol cardinal en la patogenia de las alteraciones metabólicas, el incremento del riesgo de enfermedad ateroesclerótica y las complicaciones tardías del síndrome.
La insulina actúa por estímulo directo en la esteroidogénesis y la foliculogénesis o de forma indirecta, potenciando la acción de la LH o favoreciendo la disfunción neuroendocrina.9,50 A nivel ovárico actúa a través de sus receptores propios o los de IGF que, por su similitud estructural, reconocen indistintamente ambas moléculas. El hiperinsulinismo incrementa la síntesis de IGF-1, el efecto de la FSH en la síntesis de receptores de LH en la granulosa y, en sinergia con la LH, promueve la actividad de la enzima citocromo P450-17alfa, la luteinización de la granulosa y la proliferación de las células de la teca y el intersticio. Funciona como una co-gonadotropina que modula la esteroidogénesis inducida por LH, causa hiperandrogenismo y favorece la anovulación. También puede alterar la expresión de genes vinculados al proceso meiótico del ovocito y afectar su calidad.9,47
El exceso de insulina aumenta la liberación hipofisaria de LH basal y estimulada por GnRH, reduce la síntesis hepática de la globulina transportadora de hormonas sexuales (SHBG) y de IGF-BP (lo que condiciona aumento de la fracción libre de los andrógenos y la IGF, con mayor actividad biológica), aumenta la síntesis de andrógenos suprarrenales mediada por ACTH y, por su acción adipogénica, favorece la obesidad y el estado pro inflamatorio. El efecto conjunto de hiperandrogenismo y obesidad empeora la RI y crea un círculo vicioso.9,47,50
Contribución de los factores ambientales
Estos factores participan en la fisiopatología del SOP por varias vías provocando cambios epigenéticos, favoreciendo o exacerbando la obesidad, RI o disfunción neuroendocrina, induciendo disminución de SHBG y generando estrés oxidativo, estado pro inflamatorio o incremento de los niveles séricos de AGE.5,6,7,8,57
La obesidad, aunque parece tener poca relevancia como factor primario en las formas típicas del SOP, se reconoce su importancia en las formas no típicas y como agravante en las formas típicas. El exceso de tejido graso contribuye a la síntesis de andrógenos a partir de formas débiles circulantes, la disminución en la síntesis hepática de SHBG, el desarrollo de hiperandrogenismo funcional adrenal y la producción de adipocinas que intervienen en la disfunción endocrina y metabólica.5-9,43) El sobrepeso de la madre en el embarazo es factor de riesgo para el desarrollo de SOP en la descendencia y para aborto, diabetes gestacional, hipertensión inducida por el embarazo, macrosomía o bajo peso al nacer.58
El Bisfenol A, un factor que ha cobrado importancia creciente por su amplio empleo en artículos de uso común y su elevado potencial para la exposición humana59) puede propiciar SOP ya que por su estructura fenólica se une al receptor de estrógenos y a SHGB, lo que incrementa la testosterona libre y la síntesis de andrógenos en la teca. Además, activa la transcripción de factores adipogénicos y sobreregula genes que estimulan la diferenciación del adipocito y la acumulación de lípidos, con lo que favorece la obesidad y la RI. Promueve un estado pro inflamatorio por inhibición de la producción de adiponectina y estímulo de la de IL-6 y TNF-α. La exposición intrauterina o neonatal se asocia a aumento de los pulsos de secreción de GnRH en la adultez y desarrollo de SOP.60
La deficiencia de vitamina D, un hallazgo frecuente en mujeres con SOP, puede contribuir al síndrome porque condiciona RI, obesidad y alteraciones metabólicas y se asocia a incremento del acúmulo de AGE en las células foliculares y a alteración de las vías intracelulares que regulan el receptor de AMH y de FSH. Su suplementación mejora la irregularidad menstrual, el desarrollo folicular y las tasas de embarazos, lo que puede considerarse una evidencia indirecta.7,8,61
Hiperandrogenismo en el SOP
El exceso de actividad androgénica es la alteración hormonal más relevante en el SOP. Depende del aumento en su producción ovárica, suprarrenal y/o periférica, del incremento en la fracción biológicamente más activa (por la disminución de SHBG que deriva del hiperandrogenismo, la disminución relativa de estrógenos, la obesidad y/o la RI) y la disminución en la actividad de las aromatasas (que hace que la conversión a estradiol sea insuficiente para lograr el aclaramiento del exceso de androstenediona producida en la teca).6,7,8,36
El hiperandrogenismo ovárico resulta del efecto conjunto de la disfunciónde las células de la teca y su aumento en número. Lo primero determina mayor secreción basal de andrógenos (por la hiperactividad enzimática) y en respuesta a la estimulación con LH (por incremento de la sensibilidad a esta). El engrosamiento de la teca, dependiente de la hipersecreción de LH, polimorfismos genéticos que codifican una LH con mayor actividad biológica, la hiperinsulinemia y el efecto modulador de factores intraováricos (IGF-1, TNFα, TGB β y otros) determina que esta hiperproducción se multiplique.36,37,62
La hiperandrogenemia se manifiesta por niveles séricos elevados de testosterona total, testosterona libre y/o androstenediona. En ocasiones existe incremento del sulfato de dihidroepiandrosterona (DHEAs), que sugiere contribución suprarrenal. Se presenta con amplias variaciones interindividuales en cuanto a intensidad, tipo y número de andrógenos elevados. En algunos casos puede no detectarse, lo que se explica por la disminución en los niveles de SHBG, que determina menor concentración de formas inactivas y enmascara la magnitud del exceso.9,36,37,63
Alteraciones de la foliculogénesis
La foliculogénesis alterada es de los elementos fisiopatogénicos de mayor impacto en el SOP y el de más difícil comprensión por su extraordinaria complejidad y porque la evidencia es muy cambiante. El mecanismo específico permanece impreciso, pero se reconoce que intervienen variantes genéticas que condicionan disfunción intrínseca de las células de la granulosa, la teca o el ovocito. También participan diversos factores intra y extraováricos que mediante interacciones endocrinas, paracrinas y autocrinas promueven un microambiente folicular que propicia anomalías en la foliculogénesis y la ovogénesis.5,6,7,8,43
El exceso de folículos funcionales se postula que es resultado del incremento en el número de folículos reclutados, el deterioro en la selección del folículo dominante (que condiciona enlentecimiento del desarrollo) y el retardo en el proceso de muerte celular (que lleva a que tengan una sobrevida prolongada). La detención de la maduración y selección folicular se atribuye a una diferenciación prematura de los folículos, cuya causa aún no se entiende completamente. Se piensa que depende del exceso de andrógenos intraováricos, el aumento de la sensibilidad a LH, la resistencia relativa a FSH (intrínseca o secundaria al aumento de AMH) y el incremento en la relación intrafolicular de andrógenos/estrógenos.5,8,43
El arresto, por un tiempo se pensó que dependía de una disfunción específica de las células foliculares, pero la evidencia actual apunta a que es secundario a la alteración de la esteroidogénesis.5,8,43) La inhibición de la atresia folicular se imputa a una modificación en los mecanismos apoptóticos, ya sea por predisposición genética o por acción de factores intra o extraováricos.7,43
Entre los factores involucrados en el crecimiento y función folicular deficiente, el hiperandrogenismo intraovárico parece ser el elemento central. El exceso de andrógenos aumenta la expresión y actividad de sus propios receptores. Amplifica sus acciones y participa de forma directa o indirecta en la génesis de todas las alteraciones: la disfunción de las células de la granulosa, la luteinización prematura de la teca, el desarrollo del estroma, el incremento de la sensibilidad a la FSH, el reclutamiento folicular y el aumento de AMH inducidos por FSH, la inhibición en la selección del folículo dominante y el arresto folicular.5,7,43
En monos, la administración de testosterona a dosis altas induce el crecimiento de gran número de folículos sin que alguno llegue a dominante y las células de la granulosa tienen aumento del receptor de FSH, altos índices de actividad mitótica y bajos índices de apoptosis.64) Estudios básicos indican que los andrógenos ováricos elevados estimulan el crecimiento del folículo en etapa preantral y pueden inhibir la ovulación.55,65) En la clínica se demuestra el desarrollo de SOP secundario a otras causas de hiperandrogenismo.
La FSH parece tener un papel relevante en el aumento del reclutamiento folicular. Se ha demostrardo que las células de la granulosa de mujeres anovuladoras con SOP son hiperrespondedoras a FSH en estadios iniciales del desarrollo folicular,66) lo que hace suponer que es posible que ésta estimule el crecimiento del folículo en etapas más tempranas de lo que ocurre en condiciones normales.67) Los andrógenos estimulan el crecimiento y la maduración folicular dependiente de FSH mediante la inducción genómica y no genómica de la expresión del FSHR en las células de la granulosa.43,65) Así, el aumento de la sensibilidad a FSH depende de la disfunción intrínseca y/o del hiperandrogenismo.7,43
Se postula que la AMH podría desempeñar un rol esencial en la detención de la maduración y el deterioro en la selección folicular como contraparte de la FSH en la autorregulación fisiológica del crecimiento y la diferenciación folicular.43,68) El mecanismo para explicar el incremento de AMH en el SOP no está claro. Por un tiempo se pensó que dependía del hiperandrogenismo pero las evidencias actuales sugieren que es inducido por la FSH y que el exceso de andrógenos actúa de forma indirecta, mediante el aumento de la sensibilidad a FSH.43,69) Niveles séricos altos de AMH se relacionan con mayor número de folículos pequeños e incremento de testosterona y/o LH, niveles altos en el líquido folicular se asocian a disminución de la aromatización y la síntesis de estradiol, ovocitos inmaduros y menor tasa de fertilización.70,71 Se ha descrito un posible efecto estimulante de la AMH en la secreción de GnRH, que es independiente de los niveles séricos de andrógenos y FSH, lo que sugiere la posibilidad de que participe en el control de la secreción de LH y el círculo vicioso del SOP.72,73,74
Los estrógenos se implican como otro posible factor vinculado al deterioro en la maduración folicular por el rol que desempeñan en la progresión del folículo antral a folículo terminal. Su producción depende de la acción de las aromatasas, cuya actividad está regulada por el gen CYP19 que es estimulado por FSH75 y se inhibe por la AMH que anula la actividad inducida por FSH.76) En presencia de estradiol se suprime el efecto estimulatorio de la FSH sobre la producción de AMH, lo cual no ocurre si la producción de este último es insuficiente.43
Las complejas interrelaciones entre andrógenos, FSH, AMH y estrógenos parecen ser la clave en la vía fisiopatogénica de la foliculogénesis alterada del SOP. Para ayudar a entenderlas, Dewailly y otros43 proponen la teoría de los dos triángulos que se suceden cronológicamente, un modelo que no es exhaustivo, pero ofrece un esquema que facilita la comprensión de las vías fundamentales (Fig. 1).
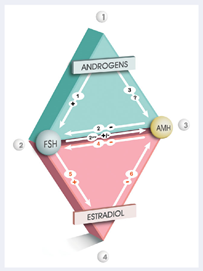
Fig. 1 Teoría de los dos triángulos para explicar las interrelaciones entre andrógenos, FSH, AMH y estrógenos.
De acuerdo con esta teoría, en condiciones fisiológicas normales en la etapa de folículo pre-antral (triángulo 1) independiente de gonadotropinas, los andrógenos incrementan la sensibilidad a la FSH. Esta última induce la producción de AMH y, dado que la secreción de estradiol es baja, la expresión de AMH es máxima, lo que disminuye la acción de la FSH, inhibe el reclutamiento folicular dependiente de ésta y protege la reserva folicular. En la etapa de folículo antral (triángulo 2) dependiente de gonadotropinas, los andrógenos disminuyen. La FSH tiene un incremento paulatino que estimula la síntesis de estradiol, el que aumenta hasta alcanzar el umbral en el que induce la supresión de la secreción de AMH y el desarrollo y maduración folicular terminal.43
Según este paradigma, en el SOP el triángulo 1 está sobredimensionado y el triángulo 2 es disfuncional. En la etapa preantral, la actividad androgénica excesiva hace que los folículos sean hipersensibles a la FSH por lo que es posible que ésta, en sinergia o potenciando la acción de factores intraováricos, estimule el crecimiento del folículo, el aumento en el reclutamiento y la producción excesiva de AMH. Durante la etapa antral, los andrógenos se mantienen elevados, la deficiencia relativa de FSH, más la inhibición de su actividad por el incremento de AMH, alteran la expresión de las aromatasas y la síntesis de estradiol, por lo que no se logra inhibir la AMH, cuyo efecto protector se exagera y lleva a la consiguiente detención en el crecimiento y la diferenciación del folículo.43
Entre otros factores, se le concede importancia a la LH. Por su papel en la hiperproducción de andrógenos, puede contribuir a todos los efectos mediados por éstos. La respuesta inapropiada a LH de las células de la granulosa de folículos pequeños determina su luteinización precoz que condiciona la detención del crecimiento de los folículos. El aumento de LH se asocia ademásl a incremento de inhibina B y a procesos meióticos prematuros en los ovocitos.7,8,38,43,50
Los niveles elevados de insulina circulante también se involucran como un factor de relevancia. Participan agravando el hiperandrogenismo, la disfunción de las células foliculares y las alteraciones en la calidad del ovocito. También por su contribución a la disfunción neuroendocrina y el aumento de LH, así como por sus efectos metabólicos y pro inflamatorios locales y sistémicos. La paradoja de la RI sistémica con hiperrespuesta folicular se explica porque la RI ovárica es selectiva (respeta las vías mitogénicas) y porque algunas de sus acciones en el ovario se ejercen a través de receptores de IGF1.5,8,47,41) Lo dicho sobre que la RI altera los procesos reproductivos solo si existe una predisposición genética previa responde el por qué no todas las mujeres con hiperinsulinemia son hiperandrogénicas.47
La producción anormal de péptidos vinculados al control intraovárico se relaciona con exceso de andrógenos, crecimiento folicular lento, alteraciones en la calidad ovocitaria y/o disfunción ovulatoria.5,7,8,77 Los IGF provenientes de la granulosa son potentes estimuladores del crecimiento folicular, mientras que el aumento del IGF-1 secretado por la teca induce detención del crecimiento.78) El incremento de EGF en el líquido folicular inhibe la síntesis de estrógenos.79) El aumento de folistatina bloquea la actividad estimulatoria de la activina en el desarrollo del folículo y la maduración de los ovocitos.7 Las inhibinas suprimen la producción de FSH y estimulan la de andrógenos.70,78,79
Los niveles elevados de BMP-15 derivado de ovocitos, TNF-α, interleucinas y de especies reactivas de oxígeno en el líquido folicular, así como la disminución de GDF-9, VEGF, NGF y la acumulación de AGE o EROS en las células de la teca y la granulosa están estrechamente asociados a anomalías del desarrollo de los ovocitos y alteraciones de la foliculogénesis.67,70,78,79,80) El incremento de las BMP procedentes de la teca estimula la secreción de AMH.81) Más recientemente se sugiere un papel potencial de las kisspeptinas producidas localmente, de biomoléculas relacionadas con el metabolismo de los carbohidratos, los lípidos o las proteínas y del aumento de homocisteína.7
Sobre la inhibición de la atresia folicular en otro tiempo se especuló que los genes de “sobrevivencia” bcl2 y mdm2 están sobrexpresados en las células de la granulosa de mujeres con SOP.82) Hoy día se propone que depende de reguladores intraováricos con funciones antiapoptóticas como el aumento intrafolicular de AMH, TNF-α, proteínas de membrana Fas o chaperonas moleculares como las proteínas de choque térmico.7,78,83
Relación con las manifestaciones clínicas
En este sentido se puede señalar en primer lugar que la gran variabilidad en la expresión clínica del SOP se corresponde con la falta de uniformidad en la presencia de los factores involucrados en la etiopatogenia del síndrome que, es heterogénea.5,6,7,8,9 Esto permite explicar la variedad de fenotipos, la ausencia de elementos distintivos del SOP en algunas pacientes y las particularidades que se observan en la respuesta terapéutica.84,85
El hiperandrogenismo clínico depende de la magnitud de la hiperandrogenemia, el tipo de andrógeno predominante y el grado de actividad biológica de estos. No siempre es proporcional a los niveles de andrógenos circulantes, lo que se imputa a anomalías genéticas en el receptor de andrógenos que disminuyen su actividad, variaciones en los niveles de SHBG o influencias étnicas.5,7) La pubarquia prematura es más frecuente cuando existe un componente adrenal en el origen del SOP o RI y sobrepeso.7,85) Se señala como elemento de importancia terapéutica que el hiperandrogenismo del SOP es funcional y dependiente de LH, por lo que cualquier intervención que suprima ésta puede suprimirlo.5
La presencia de hiperprolactinemia en el SOP no tiene una definición clara, aunque se ha intentado explicar de diferentes maneras. En teoría, el SOP puede condicionar hiperproducción de prolactina por el efecto estimulatorio de los estrógenos circulantes crónicamente elevados o la disminución del tono dopaminérgico del eje hipotálamo-hipofisario, pero esto no ha podido probarse consistentemente.33) Al mismo tiempo, la hiperprolactinemia puede inducir un SOP secundario por sus efectos en la síntesis de andrógenos y la secreción de gonadotropinas, lo cual se señala como un hallazgo frecuente y un mecanismo más probable.86
Las alteraciones en la foliculogénesis provocan hiperandrogenismo, anovulación y morfología ovárica poliquística (MOP). Se expresan clínicamente por hirsutismo, acné, alopecia androgénica, trastornos menstruales, infertilidad, riesgo de aborto, síndrome de hiperestimulación ovárica (SHEO) o resistencia a la inducción de ovulación.5) El riesgo de hiperplasia y carcinoma endometrial se relaciona con la obesidad, el hiperandrogenismo, el hiperestrinismo relativo y la infertilidad.7
La MOP no es patognomónica del SOP, se observa en otros trastornos que cursan con hiperandrogenismo y en alrededor del 30 % de las mujeres con función ovárica normal.87) Su frecuencia es mayor en adolescentes y disminuye en la mediana edad.87) Algunos autores opinan que cuando se presenta de forma aislada puede tratarse de una variante menor del espectro fenotípico del síndrome o una etapa temprana en el continuo natural del SOP.47) El número de folículos observables por ecografía obedece a la cantidad de folículos funcionales y es proporcional a las concentraciones séricas de AMH.88 El aumento del volumen ovárico se explica por el exceso de folículos y el engrosamiento del estroma.89
La infertilidad depende, además de la anovulación, de las anomalías en la calidad de los ovocitos y alteraciones en el funcionamiento y receptividad del endometrio, las que se atribuyen a trastornos genéticos o al hiperandrogenismo, el exceso de LH, la secreción de péptidos locales, la RI y/o la obesidad.22,77,90) El riesgo de desarrollar SHEO o resistencia a la inducción de la ovulación se imputa a la respuesta folicular aberrante a gonadotropinas y a variaciones en el tono de la AMH.43) Para el SHEO se postula que el aumento suprafisiológico de FSH derivado de la administración exógena supera el efecto inhibitorio de la AMH, y los folículos, por el incremento intrínseco en el número y la sensibilidad de los FSHR.43) También se señala que pueden presentarse polimorfismos en genes que determinan reducción de la unión de la AMH a sus receptores y condicionan mayor sensibilidad folicular a la FSH.91 La resistencia a la inducción de la ovulación es mayor en pacientes con hipertecosis (variante grave del SOP), RI severa o niveles séricos persistentemente elevados de LH.50
El riesgo aumentado de aborto no tiene una etiología clara. El sobrepeso corporal es un factor de riesgo bien establecido y se sugiere la posible contribución de las alteraciones en la calidad y función del ovocito, la disminución en la expresión de moléculas de implantación como la integrina α y la gliccodelina o alteraciones de factores locales derivadas del hiperandrogenismo y/o RI.7,92) Las complicaciones gestacionales se relacionan con el hiperandrogenismo materno y placentario, niveles elevados de AMH durante el embarazo, RI y obesidad, que condicionan alteraciones en el metabolismo de la glucosa, la vascularización uterina o aumento de citocinas proinflamatorias (IL, TNFα) en el citotrofoblasto y sincitiotrofoblasto.7,93,94) El parto prematuro y cesárea se asocian a estas complicaciones95) y el bajo peso al nacer a RI, obesidad materna o diabetes gestacional.26
La mayor prevalencia de diabetes mellitus, dislipidemias o estado proinflamatorio en mujeres con SOP se imputa a la RI, la obesidad, otros factores ambientales o a modificaciones genéticas específicas para estos trastornos.6,7,96,97 El riesgo de enfermedad ateroesclerótica se relaciona con las alteraciones anteriores.96,98 El fenotipo clásico del SOP expresa más manifestaciones metabólicas96) y las variantes metabólicas severas se asocian a peor pronóstico reproductivo.99) La pérdida de peso mejora el perfil metabólico, los trastornos menstruales, la tasa de ovulación y embarazo, las complicaciones gestacionales y perinatales.29,100
Conclusiones
La fisiopatología del SOP es tan compleja como apasionante. Muchas cuestiones permanecen sin esclarecerse, pero se tiene cada vez más conocimiento que aporta luz a los enigmas que aún persisten y a la comprensión de fenómenos previamente desconocidos. Existe la convicción creciente de que la alteración central en el origen y desarrollo del síndrome es ovárica y que la heterogeneidad en todos sus elementos es una característica distintiva del mismo.
Conocer la gran diversidad de factores y mecanismos que intervienen en su etiología y patogenia es fundamental, además, por su utilidad práctica para entender las amplias implicaciones del síndrome, su variabilidad clínica, la necesidad del manejo médico individualizado, la relevancia de la prevención de las complicaciones y como sustento teórico para elegir entre las opciones terapéuticas o perfeccionar aquellas para las que aún no existe una solución efectiva.

