










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de Ciencias Médicas de Pinar del Río
versión On-line ISSN 1561-3194
Rev Ciencias Médicas v.13 n.4 Pinar del Río oct.-dic. 2009
SOCIEDADES CIENTÍFICAS
Clinical-ophthalmologic and genetic characterization of Retinitis Pigmentosa in Pinar del Rio Province, Cuba. 2008
Felipe Acosta Rodríguez1, Mirlanea López Torres2, Jesús Juan Rodríguez3, José Carlos Moreno Domínguez4.
1 Asistente. Dr. Especialista de Primer Grado en Oftalmología. Centro Provincial de Retinosis Pigmentaria. Pinar del Río.
2 Instructor. Dra. Especialista de Primer Grado en Oftalmología. Centro Provincial de Retinosis Pigmentaria. Pinar del Río.
3 Profesor Auxiliar. Dr. Especialista de Segundo Grado en Pediatría y Genética Clínica. Centro Provincial de Retinosis Pigmentaria. Pinar del Río.
4 Profesor Auxiliar. Dr. Especialista de Segundo Grado en Oftalmología. Centro Provincial de Retinosis Pigmentaria. Pinar del Río.
RESUMEN
Introducción: La Retinosis Pigmentaria es una enfermedad crónica, correspondiente a las distrofias retinianas, de ahí su carácter hereditario, lento y progresivo, donde la función de los fotorreceptores y el epitelio pigmentario están afectados difusa y primariamente, caracterizada fundamentalmente por la pérdida de la visión periférica y nocturna; ocasiona alteraciones del campo visual y electrorretinograma subnormal o extinguido. Objetivo: Conocer los resultados de la caracterización Clínico- Oftalmológica y Genética de la Retinosis Pigmentaria en la provincia de Pinar del Río (Cuba). Método: Se realizó una investigación fundamental, aplicada, descriptiva y transversal que incluyó el universo de los 257 casos de Retinosis Pigmentaria, atendidos en el Hospital "III Congreso" de Pinar del Río, en el periodo comprendido desde diciembre de 1992 hasta diciembre de 2008. Resultados: La Tasa provincial de RP es actualmente de 3.51/ 10000 habitantes para una prevalencia de 1.2845. Predominó la forma Típica (70.1%), el patrón autosómico recesivo (53.4%), debut precoz (51.8%) y el estadio I-II (64.2%). La enfermedad fue más frecuente en el sexo masculino (M/F-1.45:1) y el índice de consanguinidad promedio fue de 22.2%.
Palabras clave: enfermedades oculares, epidemiología, herencia
ABSTRACT
Introduction: Retinitis Pigmentosa (RP) is a chronic condition corresponding to the retinal dystrophies having an inherited, slow and progressive character where the function of the photoreceptors and pigmentary epithelium are affected diffuse and primarily, mainly characterized by the peripheral and nocturnal loss of vision; it provokes disorders of the visual field and subnormal extinct electroretinogram. Objective: To know the results of the Clinical-ophthalmologic and genetic characterization of Retinitis Pigmentosa in Pinar del Rio Province, Cuba. Method: A fundamental, applied, descriptive and cross-sectional research including the universe of the 257 cases suffering from Retinitis Pigmentosa attended at "Tercer Congreso" Hospital in Pinar del Rio during December 1992 to December 2008. Results: The current provincial rate of RP is 3.51/10000 inhabitants with a prevalence of 1.2845. Atypical form prevailed (70.1%), autosomic recessive pattern (53.4%), early onset (51.8%) and the stages 1-2 (64.2%). The disease was more frequent in male sex (M/F-1.45:1) and the average consanguinity rate was 22.2%.
Key words: eye diseases, epidemiology, inheritance.
INTRODUCCIÓN
La Retinosis Pigmentaria (RP) es una enfermedad crónica, correspondiente a las distrofias retinianas, de ahí su carácter hereditario, lento y progresivo, donde la función de los fotorreceptores y el epitelio pigmentario están afectados difusa y primariamente; se caracteriza fundamentalmente por la pérdida de la visión periférica y nocturna; ocasiona alteraciones del campo visual (CV) y electrorretinograma (ERG) subnormal o extinguido.1-2
Es una enfermedad hereditaria en cuyo fondo del ojo (FO) pueden observarse alteraciones a nivel de la retina, pupila y coroides que varían según el estadío clínico, encontrando en un 90% de los casos alteraciones del vítreo y cristalino.1
Subjetivamente las primeras manifestaciones clínicas que expresan los pacientes son: mala visión nocturna, trastornos de la visión en los cambios de iluminación, tropezar frecuentemente con los objetos, así como fotofobia y alteraciones de la visión.1, 3
Las distrofias retinocoroideas y entre ellas la RP constituyen la cuarta causa de ceguera en el mundo, 4-5 son enfermedades que causan grave discapacidad visual; su evolución lenta y progresiva lleva al paciente a la disminución considerable de la visión y en ocasiones a la ceguera total o parcial.2, 6- 7
Otra característica clínica a destacar es la posible asociación de RP con enfermedades generales, endocrinas, trastornos auditivos, neurológicos, etc., que conforman verdaderos síndromes. Se trata entonces de una RP Asociada.5,8
Muchos han sido los esfuerzos y las investigaciones realizadas en aras de detener el progreso de la enfermedad, siendo relevantes los trabajos de inolvidable Profesor Dr. C.Orfilio Peláez Molina, quien fue el iniciador de estas investigaciones en Cuba y precursor del Programa Nacional Cubano de pesquizaje y atención integral de estos pacientes y sus familiares.1
El 5 de diciembre de 1992 quedó inaugurado en Pinar del Río por el Profesor DrC. Peláez Molina, el Centro Provincial de Retinosis Pigmentaria, en el cual se ha venido trabajando ininterrumpidamente desde entonces, por estas razones se realizó este trabajo, con el objetivo de conocer los resultados de la caracterización Clínico- Oftalmológica y Genética de la Retinosis Pigmentaria en Pinar del Río.
MÉTODOS
Se realizó una investigación fundamental, aplicada, descriptiva y transversal que incluyó el universo de los 257 pacientes con el diagnostico de RP, atendidos en el Centro Provincial de Retinosis Pigmentaria, situado en el Hospital "III Congreso" de la ciudad de Pinar del Río Cuba), desde diciembre de 1992 hasta diciembre de 2008.
En el archivo de dicho Hospital se encuentran las Historias Clínicas (HC) de todos los pacientes en las cuales está reflejada el examen oftalmológico completo que incluye: biomicroscopía del segmento anterior y posterior, agudeza visual (AV), refracción y pericampimetría. Se realiza en cada ingreso con el objetivo de conocer la evolución y/o progresión de la enfermedad, así como la respuesta al tratamiento quirúrgico y médico con el ozono y las vitaminas. Todos los pacientes tienen realizado ERG y han sido valorados en consulta de Genética Clínica, y algunos por otras especialidades médicas cuando así lo han requerido.
Partiendo de los datos reflejados en las HC, se realizó una encuesta que incluyó: nombres y apellidos, edad, sexo, raza, forma clínica de la enfermedad, edad de debut, tipo de herencia y estadío clínico del paciente en el momento de la investigación, basados en la Clasificación Cubana de la RP del DrC. Orfilio Peláez Molina (Experiencia Cubana en la Retinosis Pigmentaria. Primer Simposio Internacional de RP, La Habana, 1994).1
A. Criterios de inclusión:
-Todos los pacientes diagnosticados y tratados en el Centro Provincial de Retinosis Pigmentaria de Pinar del Río por presentar RP.
B. Criterios de exclusión:
- Pacientes con diagnostico de RP fallecidos.
- Pacientes que no deseen participar en la investigación.
- El universo y la muestra estuvieron constituidos por 257 pacientes.
- Estos datos fueron reflejados en las tablas y procesados por el método porcentual.
RESULTADOS
El estudio realizado en la provincia de Pinar del Río (Cuba), reflejó los siguientes resultados.
La Tasa provincial de RP es actualmente de 3.51/10000 habitantes para una prevalencia de 1:2845.
Del total de pacientes con RP, el 70.1% presentan la forma Típica de la enfermedad, son Asociadas el 20.2% y sólo el 9.7% son Atípicas, la herencia autosómica recesiva es la forma más frecuente, representada en un 63.4% de los pacientes (Tabla1).
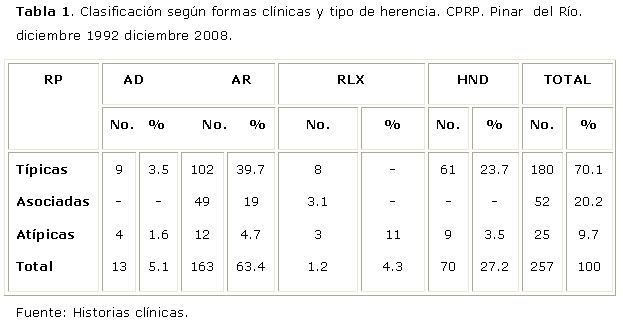
Leyenda
AD: Autosómica dominante.
AR: Autosómica recesiva.
RLX: Recesiva ligada al cromosoma X.
HND: Herencia No Definida.
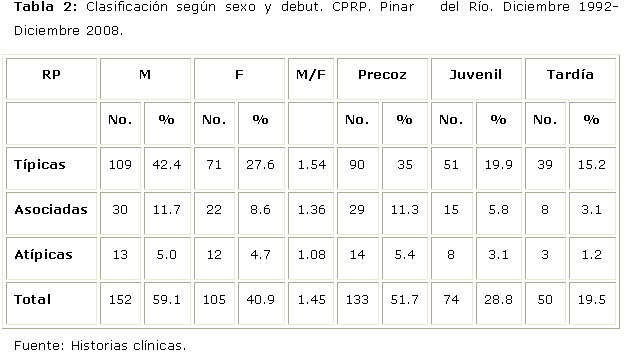
El sexo masculino (M/F=1.45:1) y el debut precoz (51.7%) predominaron en todas las formas clínicas (Tabla 2).
El estadío I-II predomina en todas las formas clínicas de la enfermedad (64.2%), el 9.7% del pacientes se encuentra en estadío III y un 26.1% en estadío IV (Tabla 3).

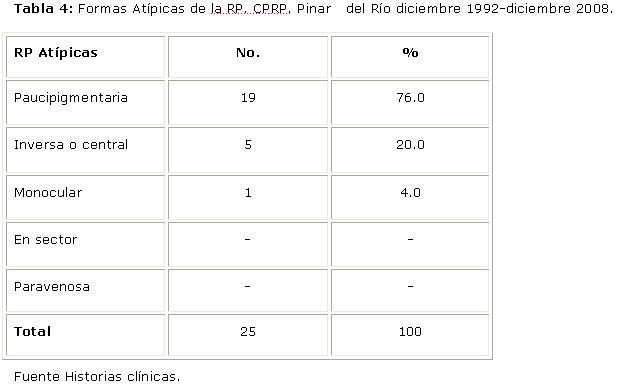
Entre las RP Atípicas, la forma paucipigmentaria representó el 76% (Tabla 4).
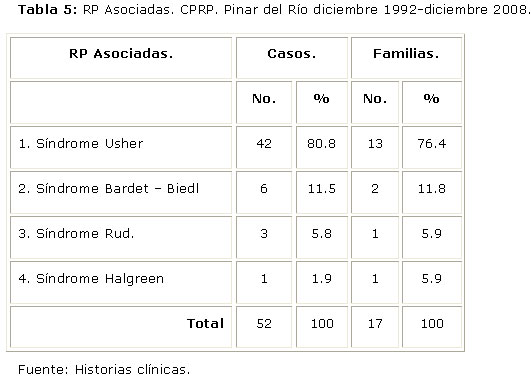
Entre las formas Asociadas, la más frecuente fue el Síndrome Usher (80.8%) (Tabla 5).
DISCUSIÓN
La Retinosis Pigmentaria afecta a 1.5 000 de seres humanos, varía de unos países a otros, en Inglaterra y España, más de 6 % de las personas ciegas menores de 65 años, deben su discapacidad visual a esa afección y se considera la primera causa de ceguera de tipo distrófico y hereditaria en el mundo.3,5
Atendiendo a las formas clínicas de la enfermedad, la forma típica fue la más frecuente en el estudio realizado en Pinar del Río, resultados que coinciden con los de las provincias cubanas: Santiago de Cuba (4), Villa Clara (9) y la Habana (10).
En cuanto a las formas de transmisión de la enfermedad, estudios cubanos realizados en las familias de la Habana y en Las Tunas,2,10 así como otros realizados en EEUU, Francia, Japón y Dinamarca8 reflejan que la herencia autosómica recesiva es la forma más frecuente de transmisión en pacientes que padecen de RP.
La RP recesiva ligada al X, representada en un 4.3% de la muestra es considerada la forma más severa de RP, con debut de la ceguera nocturna antes de los 20 años de edad, agudeza visual disminuida de manera marcada en edades tempranas y ERG subnormal o extinguido.3,11 Se destacó que no siempre los enfermos de las familias con RP recesiva ligada al X han tenido la peor evolución hacia la ceguera como expresa la literatura hasta el momento; esto coincide con otros estudios realizados en Cuba10 y en el mundo.12
Según la Clasificación Cubana de la RP del Dr.C. Orfilio Peláez Molina (Experiencia Cubana en la Retinosis Pigmentaria. Primer Simposio Internacional de RP, La Habana, 1994)1, podemos decir que la enfermedad teniendo en cuenta la edad se clasifica en:
· Comienzo precoz: cuando esta se manifiesta antes de los 10 años de edad
· Comienzo juvenil: entre los 10-20 años
· Comienzo tardío: después de los 21 años.
Diversos autores señalan que el debut de la enfermedad ha sido siempre antes de los 20 años, agregan además que el comienzo por encima de los 50 años es excepcional.1 El debut precoz y el sexo masculino son resultados también expresados en los estudios de otros autores cubanos.5, 9-10, 13-14
La forma pauciforme de la enfermedad se describe en aquellos pacientes con síntomas y signos típicos pero que oftalmológicamente no presentan pigmentos en el fondo de ojo. Esta ha sido la forma de presentación de RP Atípica más frecuente en el estudio, y coincide con los resultados encontrados en los estudios realizados en las provincias cubanas de Las Tunas, Villa Clara, Camagüey y La Habana.2,9-10, 13-14
En ocasiones la RP forma parte de un síndrome (RP Asociada) y no aparece el cortejo sintomático que hace pensar en esta enfermedad inicialmente (1). El Síndrome Usher es la forma más frecuente de RP Asociada, constituye la primera causa de sordo ceguera hereditaria y su prevalencia es variable. Oscila entre 3,0 / 100 000 en Escandinavia y 4,4 / 100 000 en Estados Unidos y en Cuba es la forma sindrómica de RP más frecuente.14
CONCLUSIONES
· La Tasa provincial de RP es actualmente de 3.51/ 10000 hab. para una prevalencia de
1.2845, predominando la forma Típica de la enfermedad (70.1%), el patrón de herencia autosómico recesivo (63.4%), el debut precoz (51.7%), el estadio I-II (64.2%) y el sexo masculino.
· En las RP Atípicas, la forma paucipigmentaria (76%) y en las RP Asociadas el Síndrome Usher (80.8%) fueron las formas clínicas más frecuentes.
REFERENCIAS BIBLIOGRÁFICAS
1- Peláez MO. Retinosis Pigmentaria. Experiencia cubana. 1 ed. La Habana: Edit. Científico-Técnica; 1997:26-29.
2- González Hess L, Ramírez Pérez E, Pérez Guerrero RM, Abreu Leyva A. Rasgos epidemiológicos de ciegos y débiles visuales por Retinosis Pigmentaria en la provincia de las Tunas. Rev Cub Oft Ene-Jun. 2003; 16(1): 22-26.
3-Ramírez Castro T, Lorenzo González ME, Hernández Baguer R. Retinosis Pigmentaria con herencia recesiva ligada al cromosoma x. Caracterización oftalmológica. Rev Cub Oft Jul-Dic. 2003; 16(2): 18-23.
4- García Espinosa SM, Freyre Luque R, Castillo Vázquez C, Navarro Scott M, Dáger Salomón M. Consideraciones oftalmológicas y genéticas sobre la asociación de Retinosis Pigmentaria con glaucoma. Medisan. 2007; 11(2): 13-15.
5- Hernández Baguer R, Copello Noblet M, Cabezas García AM, Domínguez Rodríguez D, Cid Vázquez B. Características clínicas y evolución de la Retinosis Pigmentaria en adolescentes. Rev Cubana Oftalmol jul.-dic. 2007; 20(2):11-15.
6- Kanski J. Oftalmología Clínica. Elsevier, Madrid; 2004.
7- Bellous AR, Kass MA, Lichtenstein SB. Retina and vitreous basic and clinical science course. Section 12. San Francisco: American Academy of Ophthalmology. 2000; 01:56-7.
8- Gutiérrez Torres SM. Retinosis Pigmentaria. Clasificación y Tratamiento. [Serie en Internet] 2007[Consultado julio 2007] Disponible en: http://retinosis.org/
9-Taboada Lugo N, Rangel Fleitas R, Membredes Pérez G. Estudio heredofamiliar de la Retinosis Pigmentaria en Villa Clara. Medicentro En-Mar.2004; 8(1):7-10.
10- Hernández Baguer R, Copello Noblet M, Dyce Gordon B. Retinosis Pigmentaria: clínica, genètica y epidemiología en estudio de familias habaneras. Rev Haban Cienc Méd Ene-Mar.2008;(8):12-15.
11-Dyce Gordon E, Mapolón Arcendor Y, Dyce Gordon B, et al. Herencia de la Retinosis Pigmentaria en la provincia Camagüey. Rev Cub Oft .1999; 12(1):58-62.
12-Dyce Gordon B, Mejías Márquez J, Copello Noblet M, et al. Aspectos genéticos y clínicos del Síndrome Usher. Rev Cub Oft. 2000; 13(2):79-83.
13-Vaughan D, Asbury T. Oftalmología General 11ed. México DF, El Manual Moderno; 2002.
14- Weleber R. G, Gregory-Evans K. Retinitis Pigmentosa and allied disorders .In Ryan S.J. Editors. Elsevier Inc. Philadelphia; 2006.
Recibido: 10 de Marzo de 2009.
Aprobado: 3 de Septiembre de 2009.
Felipe Acosta Rodríguez. Centro Provincial de Retinosis Pigmentaria. Pinar del Río. Cuba, E-mail:mirlafel@princesa.pri.sld.cu

