










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de Ciencias Médicas de Pinar del Río
versión On-line ISSN 1561-3194
Rev Ciencias Médicas v.15 n.2 Pinar del Río abr.-jun. 2011
Síndrome de Waardenburg. Variabilidad en una familia en Sandino, Pinar del Río, Cuba
Waardenburg syndrome: variability in a Sandino family
Fidel Castro Pérez1, Yenia Ledesma Vega2, Carlos Ivis Otaño Placencia3, Pedro Antonio Ramírez Sosa4, Milagros Ramos Cruz5.
1Especialista de Segundo Grado en Otorrinolaringología. Profesor Auxiliar. Policlínico Dr. Ernesto Guevara de la Serna.
E-mail: fcastro@princesa.pri.sld.cu
2Especialista de Primer Grado en Genética Médica. Policlínico Dr. Ernesto Guevara de la Serna. E-mail: yenia2009@princesa.pri.sld.cu
3Especialista de Primer Grado en Medicina General Integral. Máster en Asesoramiento Genético. Policlínico Juan Navarro.
E-Mail: carlvis@princesa.pri.sld.cu
4 Especialista de primer Grado en Medicina General Integral. Máster en Asesoramiento Genético. Instructor. Policlínico Dr. Ernesto Guevara de la Serna. E-mail: pedgenet@princesa.pri.sld.cu.
5 Especialista de Primer Grado en Otorrinolaringología. Profesor Auxiliar. Hospital Pediátrico Provincial Pepe Portilla.
E-Mail: milagros@princesa.pri.sld.cu
RESUMEN
El síndrome de Waardenburg es una enfermedad infrecuente, autosómica y dominante que cursa con cierto grado de discapacidad cuando aparece la hipoacusia neurosensorial, sin embargo, se ha detectado en Sandino, Pinar del Río, Cuba, una familia con 26 individuos vivos portadores del síndrome. Se describe por primera vez el estudio general de la familia desde el primer portador, y se demuestra la variabilidad individual en los portadores.
DeCS: SÍNDROME DE WAARDENBURG/genética; SÍNDROME DE WAARDENBURG/fisiopatología; IRIS; SORDERA; Pigmentación.
ABSTRACT
Waardenburg Syndrome (WS) is a non-frequent autosomal dominant disease, showing a certain degree of disability when neurosensitive hypoacusia appears; however, in Sandino, Pinar de Río, Cuba a family, from whom 26 alive relatives show WS. The general study of this family from the first ill relative, and the individual variability is showed.
DeCS: Waardenburg syndrome/genetics; Waardenburg, syndrome/physiopathology; iris, deafness, pigmentation.
INTRODUCCIÓN
El síndrome de Waardenburg lleva su nombre en honor a su descubridor, el oftalmólogo y genetista holandés Petrus Johannes Waardenburg, quien lo describió inicialmente en 1951. Se hereda como un rasgo autosómico dominante. Lo que significa que es suficiente con el gen de solo uno de los padres para que el niño resulte afectado1.
Los criterios diagnósticos del síndrome de Waardenburg son clínicos. Considerándose válido el diagnóstico cuando existen dos criterios mayores o uno mayor y dos menores. Los criterios mayores son: sordera neurosensorial congénita, alteraciones pigmentarias del iris, hipopigmentación del cabello (mechón de cabello blanco), familiar de primer grado afectado y distopia cantorum. Los criterios menores son: manchas cutáneas hipopigmentarias, sinofidria, raíz nasal ancha, orificios nasales antevertidos, poliosis y filtro labial corto2.
El síndrome de Waardenburg se clasifica en diferentes grados o tipos, según los síntomas presentes en el paciente.Existen 4 variantes de la enfermedad de Waardenburg3:
Tipo I: presenta todas las manifestaciones faciales y visuales que caracterizan el síndrome. El 25 % presentan sordera neurosensorial y la mayoría distopia cantorum.
Tipo II: no presentan distopia cantorum, más del 70 % presentan sordera neurosensorial y el resto de alteraciones faciales y visuales son menos frecuentes.4
Tipo III o Waardenburg-Klein: es similar al tipo I asociando alteraciones musculoesqueléticas y en algunos pacientes retraso mental y microcefalia.
Tipo IV o Waardenburg-Shah: asocia las características del síndrome de Waardenburg a megacolon congénito agangliónico (enfermedad de Hirschsprung).5
Otras malformaciones que pueden aparecer asociadas a las características de este síndrome son: meningocele, alteraciones cardíacas, atresia anal y malformaciones uterinas y vaginales. En el caso presentado existía una comunicación interventricular, y aunque no se han encontrado otros casos descritos en la literatura especializada, hipospadias peneana y criptorquidia bilateral.
La incidencia del síndrome de waardenburg en de 1/270.000 nacidos vivos, aunque debido a su baja penetrancia (20 %) la frecuencia total estimada es de1/42.000 personas. 1
Este síndrome es además el causante de hasta el 3% de los casos de sordera neurosensorial. De los cuatro tipos descritos el tipo IV es el más infrecuente del que solamente se han reportado unos treinta casos. Es, además, el causante de hasta el 3 % de los casos de sordera neurosensorial. De los 4 tipos descritos, el tipo IV el más infrecuente existiendo en la literatura especializada unos 50 casos descritos.6
Los grupos municipales de genética médica creados para la atención de cualquier discapacidad de origen genético y con el logro de una elevada formación profesional, han brindado un considerable asesoramiento para nuestra formación integral, ocupan un punto importante en la atención secuencial, lógica, de profundidad creciente de los pacientes con cualquier discapacidad de esta condición.
Constituye, por tanto, el objetivo fundamental del presente trabajo realizar una presentación general de la familia portadora del síndrome de Waardenburg, como paso inicial de una investigación lógica y secuencial, que permitirá conocer todos los aspectos posibles a investigar de una manera objetiva y con la profundad requerida y analizar hasta donde es posible hacer con la investigación, con vista a lograr un mayor aprovechamiento de las potencialidades, una vez conocidas sus principales características y peculiaridades.
MÉTODO
Se realizaron varios estudios de casos, método transversal, descriptivo, cuantitativo y observacional, a las personas pertenecientes a una familia con síndrome de Waardenburg del municipio Sandino. Se confeccionó una base datos, automatizada, con todas las variables de interés, que incluyeron características faciales, de la piel y las faneras, las auditivas y las oculares. Se utilizaron las frecuencias absolutas y relativas porcentuales para resumir las variables cualitativas; estas se compararon mediante la prueba de X2 al 95 % de certeza.
RESULTADOS
PRESENTACIÓN DE LA FAMILIA.
Se trata a una familia residente en las localidades de El Carril, poblado de Manuel Lazo y Ciudad Sandino, pertenecientes al municipio Sandino, provincia Pinar del Río, Cuba. Esta familia cuenta con varios miembros afectados por la enfermedad, la cual se ha presentado en seis generaciones estudiadas (Fig. 1). Treinta y ocho personas han presentado el síndrome, de los cuales se encuentran vivos 26.
Se han recogido en el árbol genealógico todos los miembros de la familia, con el síndrome o sin él. Para ejemplificar el predominio dominante de la enfermedad, observe en el árbol genealógico de esta familia (Fig. 1).


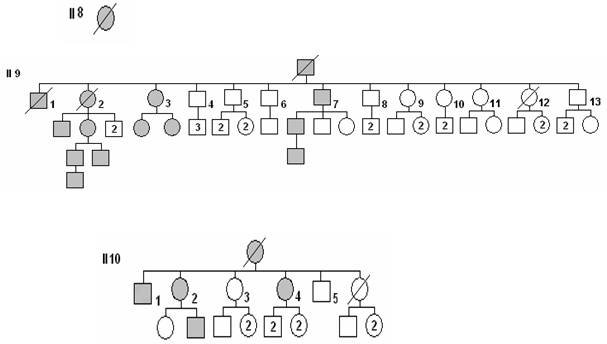
Fig. 1. Árbol genealógico por seis generaciones.

El total de miembros de esta familia, según se muestra en el árbol genealógico (Fig. 1) es de 221 personas, de los cuales hay 26 actualmente vivos con el síndrome (11,7 % del total), 14 masculinos (58.3 %) y 12 femeninos (47.2 %), que no muestran diferencias significativas en cuanto al sexo. (X2 = 0.08; gdl =1; p = 0.78).
Esta afección cursa con diversidad de expresión y existen personas con hipoacusia que puede ser parcial o total.
DISCUSIÓN
Del total de 221 miembros de la familia vivos, 26 pacientes presentan el síndrome, lo que representa el 11,7%. Si se tiene en cuenta lo infrecuente según la literatura3, se puede decir que es una muestra considerable.
Este síndrome, es además, el causante de hasta el 3% de los casos de sordera neurosensorial. De los cuatro tipos descritos, el tipo IV es el más infrecuente existiendo en la literatura especializada unos 50 casos descritos.6
Con relación al sexo la no diferencia de frecuencias del síndrome se puede explicar porque se transmite por un patrón de herencia autosómico dominante. Los 26 individuos presentan rasgos dismórficos, pero no uniformemente distribuidos en todos, por lo que se necesita investigación más profunda para particularizar en las características propias de cada caso.3, 6-10
En 18 individuos se detectaron mechones blancos en el cabello, rasgo común en muchos casos portadores del síndrome como se ha comprobado en otros estudios.11-14 Sin embargo, en nuestra casuística se presentaba de diversas formas, en unos era solo un mechón en el cabello (poliosis), en otros estaba presenta en las cejas y el bigote, o encanecimiento prematuro, como se pudo comprobar en varios de los casos analizados.
Un total de 14 afectados con el síndrome padecen de hipoacusia de intensidad variable (53,8%). Es necesario recordar que este síndrome es el responsable del 3% aproximadamente de las hipoacusias sindrómicas.6 Se presentó en algunos unilateral y en otros bilateralmente. El grado de hipoacusia puede ser variable, pero siempre es de tipo neurosensorial, generalmente bilateral, pero en ocasiones la intensidad no es la misma de un oído a otro, puede ser moderada, severa, profunda o presentarse en forma de cofosis, existiendo tres sordomudos en nuestro estudio, por esa característica.
Se comprueba la variabilidad del síndrome en los diferentes individuos. En este estudio no fue posible clasificar las variantes genéticas del síndrome en las personas afectadas, pues se carecía de estudio del cariotipo en el momento de realizar la investigación de discapacitados en el municipio de Sandino, lo cual es un desafío para futuras investigaciones.
REFERENCIAS BIBLIOGRÁFICAS
1 - Síndrome de Waardenburg. Enciclopedia médica en español [monografía en internet]. 24 abril 2008. Disponible en: http://www.nlm.nih.gov/medlineplus/spanish/encyclopedia.html [Acceso 10 de marzo de 2011]
2. Dourmishev AL, Dourmmishev LA, Schwartz RA, Janniger CK. Waardenburg syndrome. Int J Dermatol. 1999; 38:656-63.
3. Reynolds JE, Meryer JM, Landa B, Stevens CA, Arnos KS, Israel J, et al. Analysis of variability of clinical manifestations in Waardenburg syndrome. Am J Med Genet. 1995; 57:540-7.
4. Pardono E, Van Bever Y, Van den Ende J, Havrenne PC, Iughetti P, Maestrelli S, et al. Waardenburg syndrome: clinical differentiation between types I and II. Am J Med Genet. 2003; 117A:223-5.
5. Shah K, Dalal SJ, Desai MP, Seth PN, Joshi NC, Ambani LM. White forelock, pigmentari disorder of irides, and long segment Hirschsprung disease: possible variant of Waardenburg syndrome. J Pediatr. 1981; 99; 432-5.
6. Abril Molina A, González Carretero A, Miras Baldó MJa, Narbona López E. Alteraciones de la pigmentación cutánea asociadas a obstrucción intestinal. Servicio de Neonatología. Hospital Clínico Universitario San Cecilio. Granada. España. An Pediatr (Barc). 2007[citado enero 2011]; 66:429-30. Disponible en: http://www.elsevier.es/en/node/2053372
7. Sznajer Y, Coldéa C, Meire F, Delierre I, Sekhara T, Touraine RL. A de novo XOS10 mutation causing severe type 4 Waardenburg suydrome without Hirschsprung disease. Amer J Hum Genet. 15 April 2008[citado enero 2011]; 146A (8): 1038-41. Disponible en: http://onlinelibrary.wiley.com/doi/10.1002 /ajmg.a.32247/full
8. arrer LA, Grundfast KM, Amos J, Arnos KS, Asher JH, Beighton P, Diehl SR, Fex J, Foy C, Friedman TB. et al. Waardenburg syndrome (WS) type I is caused by defects at multiple loci, one of which is near ALPP on chromosome 2: first report of the WS consortium. Amer J Hum Genet. 1992[citado enero 2011]; 50: 902-13. Disponible en: http://www.nih.gov
9. Tamayo ML, Gelvez N, Rodriguez M, Florez S, Varon C, Medina D, Bernal JE. Screening program for Waardenburg syndrome in Colombia: clinical definition and phenotypic variability. Amer J Hum Genet 15 Apr 2008[citado enero 2011]; 146A
(6): 1026-1031. Disponible en: http://http://www.ajmg.a.32189 [Acceso 04-04-2010]
10. Pardono E, Mazzeu JF, Lezirovitz K, Auricchio MT BM, Iughetti P, Nascimento RMP, Mingroni-Netto RC, Otto PA. Waardenburg Syndrome: Description of two novel mutations in the PAX3 gene, one of which incompletely penetrant. Genetic and Molecular Biology, 2006[citado enero 2011]; 29 (4): 601-04. Disponible en: http://www.scielo.br/pdf/gmb/v29n4/32106.pdf
11. Heller M, Guinle L, Airaudo C, Rodríguez EA. Vogt-Koyanagi-Harada syndrome: Association with alopecia areata. Sociedad Argentina de Dermatología [revista en internet]. 2007 [citado 10 marzo de 2011]; 14(2):193-4. Disponible en: http://sad.org.ar/revista/pdf/2007-2.pdf#page=31
12.Thapa R, Mallick D, Ghosh A, Ghosh A. Waardenburg syndrome associated with laryngomalacia. Singapore Medical Journal [revista em internet]. 2009 [citado 10 marzo de 2011]; 50(12):401. Disponible en: http://smj.sma.org.sg/5012/5012cr3.pdf
13. Tobin AM, Kirby B. Successful treatment of recalcitrant acrodermatitis continua of Hallopeau with adalimumab and acitretin. Br J Dermatol2005; 153 (2): 440-66.11-5. Disponible en: http://onlinelibrary.wiley.com/doi/10.1111/j.1365-2133.2005.06759.x/abstract
14. Adisen E, Oztas M, Gurer MA. Lack of efficacy of etanercept in acrodermatitis continua of Hallopeau. Int J Dermatol 2007[citado enero 2011]; 46 (11): 1205-07. Disponible en: http://onlinelibrary.wiley.com/doi/10.1111 /j.1365-4632.2007.03251.x/full <
Recibido: 26 de abril de 2011.
Aprobado: 3 de mayo de 2011.
Dr. Fidel Castro Pérez. Especialista de Segundo Grado en Otorrinolaringología. Profesor Auxiliar. Policlínico Dr. Ernesto Guevara de la Serna.
E-mail: fcastro@princesa.pri.sld.cu

