










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de Ciencias Médicas de Pinar del Río
versión On-line ISSN 1561-3194
Rev Ciencias Médicas v.15 n.2 Pinar del Río abr.-jun. 2011
Síndrome de Miller Fisher. A propósito de un caso
Miller Fisher syndrome. A case report
Juan Manuel Zaldívar Rodríguez1, Jackeline Sosa Hernández2, Dinaisy García Torres3, Anita de las Mercedes Guillén Canavas4, Oscar Lázaro Pérez Alfonso5.
1Especialista de Primer Grado en Medicina General Integral y en Neurología. Máster en Atención Integral al Niño. Instructor. Hospital Provincial Pediátrico Docente Pepe Portilla. E-mail: zaldivar@princesa.pri.sld.cu
2Especialista de Primer Grado en Medicina General Integral. Residente de Psiquiatría Infanto Juvenil. Máster en Atención Integral al Niño. Instructora. Hospital Provincial Pediátrico Docente Pepe Portilla. E-mail: jameli06@princesa.pri.sld.cu
3Residente de Pediatría. Hospital Provincial Pediátrico Docente Pepe Portilla. E-mail: emanuel@princesa.pri.sld.cu
4Especialista de Primer Grado en Pediatría. Instructora. Hospital Provincial Pediátrico Docente Pepe Portilla. E-mail: enevel@princesa.pri.sld.cu
5Especialista de Primer Grado en Medicina General Integral y en Pediatría. Hospital Provincial Pediátrico Docente Pepe Portilla.
RESUMEN
El síndrome de Miller Fisher es una variedad clínica del síndrome de Guillain-Barré, que se caracteriza por oftalmoplejía, arreflexia y ataxia. Se presenta el caso de una paciente de 6 años de edad, atendida en el Hospital Pediátrico Docente Pepe Portilla de Pinar del Río, en noviembre de 2010; con antecedentes de salud anterior, que 5 días previos al inicio del cuadro tuvo una infección respiratoria alta aguda, seguida de un cuadro de debilidad bucofaríngea y facial, ataxia y debilidad muscular intensa de las extremidades. Se interconsulta con el servicio de Neurología, donde se constata diplejía facial periférica, parálisis del IV par izquierdo, ataxia e hiporreflexia; se le realizan varias investigaciones e ingresa en la Unidad de Cuidados Intensivos con el diagnóstico de síndrome de Miller Fisher. Se comienza el tratamiento con terapéuticocon, intacglobin y vitaminoterapia, logrando una evolución satisfactoria de la paciente a los 10 días de su ingreso en el servicio de UCI con regresión del cuadro.
DeCS: SÍNDROME DE MILLER FISHER (SMF)/complicaciones, SÍNDROME DE GUILLAIN-BARRÉ (SGB)/diagnóstico, INFECCIÓN, EVOLUCIÓN.
ABSTRACT
Miller Fisher syndrome is a clinical variant of Guillain-Barré syndrome it is characterized by ophthalmoplegia, areflexia and ataxia. A six-year old female patient attended to "Pepe Portilla" Children Hospital, Pinar del Rio in November 2010; with past health records, who 5 days before the onset of the disorder suffered from an acute upper respiratory infection that was followed by a picture of buccopharyngeal and facial weakness, ataxia and acute muscular debility of the limbs. A referral to Neurology service verified peripheral facial diplegia, paralysis of the IV left pair, ataxia and hyporeflexia; several examinations were performed before admitting her to the Intensive Care Unit (ICU) with the diagnosis of Miller Fisher syndrome. The therapeutic treatment included intacglobin and vitamins, achieving a satisfactory progress; the patient was discharged from the ICU 10 days after her admission with a total regression of the clinical picture.
DeCS: Miller Fisher Syndrome (MFS)/complications, Gillain-Barré Syndrome (GBS)/diagnosis, infection, evolution.
INTRODUCCIÓN
El síndrome de Miller Fisher (SMF) es una variante reconocida del síndrome de Guillain Barré (SGB), descrito inicialmente por C. Miller Fisher en el año 19561 caracterizado entre otras manifestaciones por oftalmoplejía externa total, ataxia profunda y arreflexia tendinosa.2,3
Este síndrome sucede con poca frecuencia en la práctica clínica, ya que su incidencia es muy baja, generalmente ocurre entre un 2 y 5 % de casi todas las series de GBS agudos.2, 3 Están descritos cuadros asociados a infecciones respiratorias o digestivas.2, 4
PRESENTACIÓN DE CASO
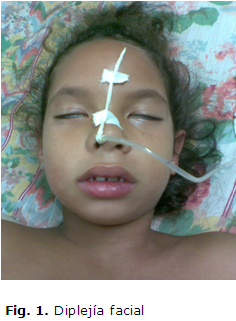
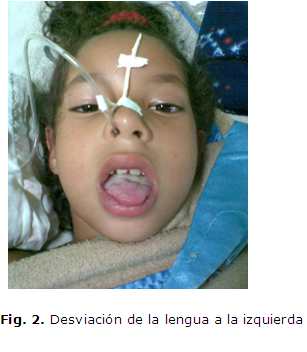
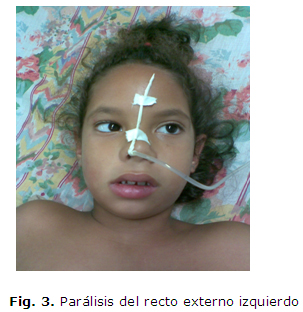
Paciente femenina de 6 años de edad, con antecedentes patológicos personales de salud. Es remitida al cuerpo de guardia del Hospital Pediátrico Docente Pepe Portilla de Pinar del Río porque 5 días previos al inicio del cuadro comenzó con manifestaciones catarrales inespecíficas, caracterizadas por tos seca, coriza y estornudos, seguido de un cuadro de instalación súbita de debilidad bucofaríngea y facial, que le dificultaba hablar y deglutir los alimentos líquidos y sólidos, con regurgitación de estos por la nariz, siendo más frecuente a los alimentos líquidos, acompañada de inestabilidad en la marcha, dificultad para cerrar los ojos y debilidad muscular intensa de las extremidades. Por tal motivo, se interconsulta el caso con el servicio de Neurología. Se le constata al examen físico neurológico una oftalmoplejía externa, con ptosis palpebral bilateral, diplejía facial periférica (fig. 1), debilidad bucofaríngea, con desviación de la lengua hacia la izquierda (fig. 2), parálisis del recto externo izquierdo (fig. 3) ataxia, aumento de la base de sustentación y trastornos cinéticos, más marcados de lado derecho, con dismetría, prueba del índice de Babinnski, del talón y de la inversión de la mano positiva, hipotonia y debilidad muscular bilateral de predominio proximal y más marcado en el miembro superior derecho. Fuerza muscular en los miembros superiores e inferiores distal de -1 a -2 y proximal de -2, más marcada a miembro superior derecho, con hiporreflexia generalizada.
Se le realizó un fondo de ojo normal, la laringoscopia indirecta y rinoscopia posterior normal, TAC de cráneo normal, RMN de cráneo sinusitis frontal, punción lumbar con una disociación albuminocitológica, con proteinoraquia de 1,9 g/l y celularidad normal. Estudios virológicos de reacción en cadena de la polimerasa (PCR) en heces fecales y LCR negativos, y un enlentecimiento de la velocidad de conducción nerviosa en miembros superiores e inferiores.
Se diagnostica el síndrome de Miller Fisher. Por tal motivo se traslada para la terapia intensiva y se le pone tratamiento con intacglobin a 400mg/kg/día durante 5 días y vitaminoterapia. Teniendo una evolución satisfactoria.
DISCUSIÓN
El SMF es un cuadro neurológico periférico (poliradiculopatía monofásica) considerado la variante más común del SGB.2-4 Habitualmente la diplopía y diparesia facial, constituyen el primer signo que se observa en el 50 % de los casos, 5 seguido varios días después por la tríada clínica clásica de oftalmoplejía, ataxia y arreflexia. La oftalmoplejía generalmente es externa y se inicia por los rectos superiores, seguido de los laterales, para finalizar en los rectos inferiores y con frecuencia se observa el fenómeno de Bell, pese a la parálisis de la mirada voluntaria.5, 6 En ocasiones, hay una toma de otros nervios craneales, como la referida por la paciente, que originan debilidad bucofaríngea y facial, con agotamiento intenso de extremidades.3, 4
El cuadro generalmente es precedido por un proceso infeccioso, sobre todo de tipo respiratorio banal o digestivo, entre un 43 y un 60% de los casos y suele estar relacionado a Campylobacter jejuni (21%), Haemophilus influenzae (8%), Citomegalovirus (3%) y Micoplasma pneumoniae (3%).2,4 El tiempo medio de aparición de los síntomas neurológicos tras la infección es de 1-2 semanas y el cuadro se desencadena por un proceso autoinmune, mediado por autoanticuerpos contra un componente de la vaina de mielina.
La etiopatogenia se basa en un fenómeno de mimetismo molecular en relación a los gangliósidos GQ1b, GD3 y GT1a, presentes en la mielina de los nervios periféricos y en las áreas paranodales de los 3°, 4° 5° y 6° pares craneales. Su diagnóstico se basa en la demostración de la seroconversión, debido a la existencia en el suero de títulos elevados de anticuerpos antigangliósidos antiGQ1b, demostrados mediante ELISA, 7,8 los que se encuentran en el 90% de los pacientes que desarrollan esta variedad. Su positividad depende del momento en que se realiza el estudio, por lo que se recomienda su investigación dentro de las primeras 4 semanas de curso clínico. La correlación entre la tríada de ataxia, arreflexia y oftalmoplejía y títulos positivos de anticuerpos anti-GQ1b confirma su especificidad para el diagnóstico del SMF. En este caso, no hubo estudios de detección de anticuerpos y la confirmación diagnóstica se sustenta en la clínica compatible y en los hallazgos paraclínicos característicos como la disociación albuminacitológica y el enlentecimiento de la velocidad de conducción nerviosa, junto con la adecuada respuesta al tratamiento con inmunoglobulinas, lo cual nos ayuda a fortalecer el diagnóstico clínico de SMF.4
El diagnóstico diferencial de esta enfermedad incluye todas las enfermedades o trastornos que pueden producir una parálisis flácida aguda, para lo cual es necesario realizar los estudios adecuados e indispensables como la resonancia magnética nuclear, que ayuda a diferenciar una supuesta polineuropatía de una mielitis o encefalomielitis, que en ocasiones, muestran una clínica similar al inicio del cuadro, período en el que el estudio del LCR, ni el neurofisiológico permiten confirmar ni descartarlo, el compromiso inicial de pares craneales oculomotores con ptosis palpebral asociada obliga a descartar miastenia gravis; la poliomielitis, aunque ya erradicada en la mayoría de los países, siempre debe considerarse, así como la inducida por vacunas o por virus no polio, como el enterovirus 71 y el virus del oeste del Nilo que se ha observado en los últimos años, también hay que tener en cuenta el botulismo y otras enfermedades neuromusculares hereditarias, como la miopatía congénita, distrofia muscular congénita o atrofia muscular espinal, no diagnosticadas previamente.9
El tratamiento de esta enfermedad es médico y de rehabilitación y comprende medidas generales y específicas, haciendo énfasis principalmente en el manejo adecuado y la preservación de las funciones respiratoria y cardiovascular, junto con el mantenimiento de hidratación y nutrición adecuadas, así como la prevención y control precoz de los procesos infecciones, que pueden agravar el curso de la enfermedad.
El tratamiento específico es el uso de inmunoglobulina en dosis de 0.4 g por kg de peso durante 5 días o de 1 g por kg de peso durante 2 días (actualmente considerada más efectiva), completando siempre una dosis total de 2 g por kg9, 10 Actualmente, se recomiendan en los casos con una progresión rápida de la debilidad muscular e insuficiencia respiratoria o necesidad de la ventilación mecánica, compromiso de pares craneales bulbares e incapacidad para deambular independientemente.9
La plasmaféresis ha mostrado igual eficacia que la inmunoglobulina pero, como es un tratamiento más invasivo y arriesgado, se reserva sólo para los casos infantiles que muestran intolerancia o que no responden a la inmunoglobulina.9, 10 La rehabilitación debe comenzar precozmente y es fundamental para lograr una recuperación más rápida e integral. El SMF tiene buen pronóstico en los niños, con una recuperación total en el 85% de los casos, la evolución es favorable y no se acompaña de una insuficiencia respiratoria ni de paresia severa de las extremidades y las recidivas son raras.5, 6 Normalmente la recuperación total es buena en un período de meses, plazo en el que desaparece la oftalmoplejía externa, sin déficit residual.7
REFERENCIAS BIBLIOGRÁFICAS
1. Fisher M. An unusual variant of acute idiopathic polyneuritis (syndrome of ophthalmoplegia, ataxia and areflexia). N Eng J Med. 1956; 255:57-65.
2. Torrent LS, Julian AN, Dueñas BP, Osorio AN, Oferil JC. Síndrome de Miller Fisher en la edad pediátrica: descripción de 3 casos.
An Pediatr Barc. 2009 oct; 71(4): 377-8.
3. Padilla-Parrado F, García-Casares N, Heras-Pérez JA, Sempere-Fernandez J, Serrano-Castro V, Romero-Acebal M. Síndrome de Miller Fisher como presentación de una encefalopatía de Wernicke. Rev Neurol. [serie en Internet] 2008 Apr. [Acceso 20 de diciembre de 2010]; 46(8): [Aprox. 3p.]. Disponible en: http://dialnet.unirioja.es/servlet/articulo?codigo=2577410
4. Cerisola-Cardoso A, Capote-Moreira G, Scavone-Mauor C. Síndrome de Guillain-Barré en pediatría. Diferentes formas de presentación y dificultades en el diagnóstico precoz. Rev Neurol. [serie en Internet] 2007. [Acceso 20 de diciembre de 2010]; 44(12): [aprox. 7p.] Disponible en: http://dialnet.unirioja.es/servlet/articulo?codigo=2327691
5. Fenichel GM. Ataxia. In: Clínical pediatric neurology. A signs and symtpoms approach. 5a. ed. Philadelphia: Elsevier Saunders; 2005.p. 219-37.
6. Torricelli RE. Síndrome de Guillain Barré en pediatría. Medicina. T-1.V-69. Buenos Aires; 2009.
7. Lestayo-O'Farrill Z, Hernández-Cáceres JL. Análisis del comportamiento del síndrome de Guillain-Barré. Consensos y discrepancias. Rev Neurol. [Serie en Internet] 2008. [Acceso 20 de diciembre de 2010]; 46(4): [Aprox. 7p.]. Disponible en: http://dialnet.unirioja.es/servlet/articulo?codigo=2550548
8. Kaida K, Kanzaki M, Morita D, Kamakura K, MotoyoshiK, Hirakawa M, et al. Anti-ganglioside complex antibodies in Miller Fisher syndrome. J Neurol Neurosurg Psychiatry. [Serie en Internet] 2006. [Acceso 20 de diciembre de 2010]; 77(9): [Aprox. 3p.] Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/16614007
9. Vilas E, Fernandez JM, Navarro C, Suárez L, García de Lorenzo A. Síndrome neuromuscular del paciente crítico. Rev Neurol. [Serie en Internet] 2006. [Acceso 20 de diciembre de 2010]; 42(11): [Aprox. 6p.] Disponible en: http://dialnet.unirioja.es/servlet/articulo?codigo=2008921
10. Agrawal S, Peake D, Whitehouse WP. Management of children with Guillain-Barré syndrome. Arch Dis Child Educ Pract Ed. [Serie en Internet] 2007. [Acceso 3 de mayo de 2008]; 92: [aprox. 7p.] Disponible en: http://ep.bmj.com/content/92/6/161.extract
Recibido: 21 de marzo de 2011.
Aprobado: 31 de marzo de 2011.
Dr. Juan Manuel Zaldívar. Especialista de Primer Grado en Medicina General Integral y en Neurología. Máster en Atención Integral al Niño. Instructor. Hospital Provincial Pediátrico Docente Pepe Portilla. E-mail: zaldivar@princesa.pri.sld.cu Dirección Particular: Justo Hidalgo No. 57 e/ Delicia y Angeles. Pinar del Río. Teléfono: 712302.


{kind=link}
{kind=link}
{kind=link}