










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de Ciencias Médicas de Pinar del Río
versión On-line ISSN 1561-3194
Rev Ciencias Médicas vol.16 no.2 Pinar del Río mar.-abr. 2012
Piebaldismo: un desorden de la pigmentación. Presentación de un caso
Piebaldism: a pigmentary disorder. A case report
Deysi Licourt Otero1, Hernán Pereda Chávez2, Yesenia Pérez Expósito3, Ledys Mabel Fernández Hernández4
1Especialista de Primer Grado en Medicina General e Integral y Segundo Grado en Genética Clínica. Asistente. Centro Provincial de Genética de Pinar del Río. Correo electrónico: deysili@princesa.pri.sld.cu
2Especialista de Primer Grado en Medicina General Integral. Máster en Asesoramiento Genético. Instructor. Policlínico Universitario "Epifanio Rojas Gil". Servicio Municipal para el desarrollo de la Genética. San Luís, Pinar del Río. Correo electrónico: peredach@princesa.pri.sld.cu
3Licenciada en Enfermería. Máster en Asesoramiento Genético. Policlínico Universitario "Epifanio Rojas Gil". Servicio Municipal para el desarrollo de la Genética. San Luís, Pinar del Río. Correo electrónico: yesepe@princesa.pri.sld.cu
4Especialista de Primer Grado en Medicina General Integral. Máster en Asesoramiento Genético. Policlínico Universitario "Epifanio Rojas Gil". Servicio Municipal para el desarrollo de la Genética. San Luís, Pinar del Río.
RESUMEN
Se presenta un niño con trastorno de la pigmentación de la piel. Posee antecedentes familiares de igual entidad. A través de la confección de la historia clínica, confección del árbol genealógico, y fundamentalmente, el examen físico se le diagnóstica de piebaldismo. Se trata de una rara entidad, por lo tanto, se decide la revisión de la literatura médica.
DeCS: PIEBALDISMO/diagnóstico; genética; etiología. ALBINISMO PARCIAL/genética; diagnóstico.
ABSTRACT
A child presenting a pigmentary disorder in the skin attended to the genetic office, the patient has a familial history with the same entity. Through the preparation of the clinical chart, family tree was examined; Piebaldism was mainly diagnosed by physical examination. Since this is a rare entity, a medical literature revision was carried out.
DeCS: PIEBALDISM/diagnosis; genetics; etiology. PARTIAL ALBINISM/genetics; diagnosis.
INTRODUCCIÓN
La pigmentación cutánea es un proceso extremadamente complejo. La coloración de la piel varía en los diferentes grupos étnicos y está influenciada por una variedad de factores como la melanina, los capilares sanguíneos, los cromóforos cutáneos (licopeno, caroteno) y el colágeno en la dermis. Otros factores físicos importantes incluyen la refracción, reflexión y absorción de la luz en la piel así como la transparencia del estrato córneo y de la epidermis. El color de la piel es controlado genéticamente por el sistema pigmentario, o sea, la melanina y los melanocitos. Los cambios anormales en la coloración de la piel son observados en un número diverso de desordenes con diferentes mecanismos de origen genético, que involucran el desarrollo en la migración de los melanoblastos (piebaldismo, Síndrome de Waanderburg, etc.), la síntesis de la melanina (albinismo oculocutáneo), y formación de melanosomas (Síndrome Hermansky-Pudlak HPS, Síndrome Chediak-Higashi CHS, Síndrome Griscelli GS).1
El piebaldismo es un desorden autosómico dominante causado por mutaciones en el gen c-kit cuyo locus se encuentra en el cromosoma 4q12-13, el cual codifica para el receptor de la tirosina quinasa necesario para la migración de los melanoblastos. Numerosas mutaciones han sido reportadas y la severidad de la expresión fenotípica se correlaciona con el sitio de la mutación dentro del gen.2
Este trabajo persigue como propósito describir en un niño las manifestaciones clínicas de piebaldismo, una enfermedad con baja frecuencia y considerada como una genodermatosis rara, el caso índice de este estudio forma parte de una familia en la que la enfermedad se está segregando con un patrón mendeliano clásico, por todo lo anterior, se decide la presentación del caso previo consentimiento informado de ambos padres para la publicación.
PRESENTACIÓN DEL CASO
Paciente de 10 años de edad, natural del municipio San Luis, provincia Pinar del Río, remitido a consulta de Genética Clínica por hiperlaxitud articular.
Historia del embarazo del caso propósito (datos relevantes):
Se realizó una captación tardía de embarazo con antecedentes de 11 años de infertilidad, hacia el tercer trimestre del embarazo se le diagnóstica por el ultrasonido oligohidramnios.
Antecedentes familiares: Se investiga a varias personas afectadas distribuidas en cinco generaciones, en las cuales predomina la hipopigmentación en el miembro inferior derecho (MID), miembro inferior izquierdo (MII) y el tronco, además del mechón de pelo blanco. Ver árbol genealógico (Figura 1)
Historia perinatal: Nace producto de un parto distócico con un tiempo gestacional de 39 semanas, con un crecimiento intrauterino retardado y apgar 9-9; no se recogen otros datos relevantes al interrogatorio. Examen físico-genético (datos positivos):
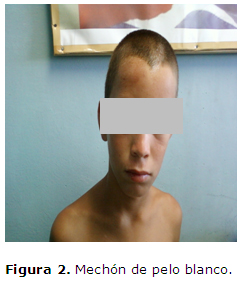
Cráneo-cara: Mechón de pelo blanco que ocupa la región paramedial izquierda que mide 6 x 3 cms. Alargada y de forma casi triangular, (Figura 2). Asimetría cráneo facial.
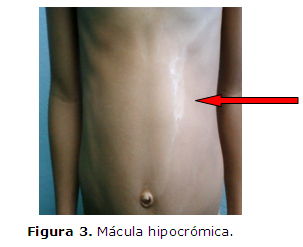
Tórax: Mácula hipocrómica que se extiende desde la región anterior izquierda y confluye hacia la línea media torácico-abdominal a nivel del ombligo y que contiene máculas hiperpigmentadas, con mácula acrómica en la región izquierda de forma lineal de 9 x 2 cms (Figura 3).
Sistema osteoarticular: Se realiza el test de Beighton para evaluar hiperlaxitud articular el cual resulta con un puntaje bajo para la misma.
Exámenes complementarios
Potencial auditivo: Sin alteraciones.
Estudios oftalmológicos: Sin alteraciones.
Diagnóstico definitivo:
Piebaldismo o albinismo parcial.
DISCUSIÓN
Las células pigmentarias derivan de una población de células pluripotentes localizadas en la cresta neural. Han sido identificados diferentes mecanismos en el desarrollo, migración y proliferación de los melanocitos; estos factores incluyen el receptor del endotelin B (EDNRB) y su ligando endothelin-3 (EDN3), así como el factor de transcripción PAX3, SOX10 y MITF entre otros. Se ha identificado otro factor que juega un rol esencial en el desarrollo de los melanocitos, el receptor KIT tipo III.3 (Figura 4)
El piebaldismo (OMIM 164920) es una enfermedad de la piel, con herencia autosómica dominante caracterizada por máculas blanquecinas en la piel, en el pelo con ausencia completa de los melanocitos, localizadas más frecuentemente en la frente, tórax, abdomen y extremidades, pueden desarrollarse máculas pigmentadas en las márgenes y dentro de las lesiones acrómicas.4
La mutación identificada en el gen KIT para el piebaldismo o albinismo parcial tiene su locus en el cromosoma 4q12; este gen codifica para una proteína tirosin kinasa. Han sido descritas más de 40 mutaciones diferentes en este gen (heteregoneidad alélica) y se han reportado los fenotipos más severos cuando la mutación en el exón 2 del gen KIT involucra el dominio intracelular tirosin kinasa produciéndose un cambio del aminoácido cisteína por glicina, y por tanto, un trastorno en la polaridad de la proteína, mientras que fenotipos más leves han sido correlacionados con mutaciones en el dominio extracelular amino-terminal.5,6
El piebaldismo ha sido descrito en todos los grupos étnicos y se distribuye con la misma frecuencia en ambos sexos. Es un trastorno congénito poco frecuente que se caracteriza por presentar un mechón blanco frontal y áreas de leucodermia (parches congénitos de piel blanca) desde el nacimiento y estables a lo largo del tiempo, como se describe en este paciente. El mechón blanco frontal, presente en el 80-90% de los pacientes, es la característica clínica más típica de este proceso. La leucodermia tiene un patrón de distribución característico, que afecta a la región anterior y central del tronco, como se aprecia en la figura 3, la porción media de las extremidades, la zona central de la frente y la región frontal media del cuero cabelludo. Las áreas de leucodermia presentan forma irregular, color blanco-lechoso, están bien circunscritas y contienen máculas hiperpigmentadas. La poliosis (mechón de pelo blanco) de las cejas y las pestañas es un hallazgo clínico frecuente.7, 8
Es necesario realizar el diagnóstico diferencial con otras entidades como el síndrome de Waanderburg tipo 1 (SW1), caracterizado por sordera o hipoacusia, distopia cantorum, alteraciones pigmentarias del íris (heterocromía total del iris, o parcial o isohipocromía (ambos ojos azules, pálidos o poço brillantes) y el cabello. Más del 90% de los individuos tienen mutaciones en el gen PAX3, lo cual evidencia la heterogeneidad de locus. El modo de herencia es autosómico dominante con una penetrancia reducida, expresividad variable y acentuada heterogeneidad genética. Otros diagnósticos que se deben tener en cuenta para diferenciar son: el vitiligo, albinismo total y el síndrome de Teitz (hipopigmentación generalizada con sordera congénita), los cuales se descartaron teniendo en cuenta el examen físico y el resultado de los exámenes auditivos y visuales realizados.9, 10
El caso integra una familia en la que hay 12 miembros afectados de piebaldismo y distribuidos en tres generaciones; en esta familia se observa la penetrancia completa, a diferencia de casos reportados en la literatura que manifiestan una penetrancia reducida y expresividad variable de la enfermedad.9,10 No se encontraron las alteraciones como la heterocromia del iris, sordera, defectos neurológicos que están descritas en otros síndromes que cursan con hipopigmentación.10
El paciente estudiado mostró los rasgos sobresalientes de la enfermedad, como son: mechón blanco triangular al momento del nacimiento y la aparición de la hipo melanosis a los pocos meses de vida, la cual adoptó el patrón típico de despigmentación, con manchas hipomelanóticas ya descritas.
REFERENCIAS BIBLIOGRÁFICAS
1. Dessinioti C, Stratigos AJ, Rigopoulos D, Katsambas AD. A review of genetic disorders of hypopigmentation: lessons learned from the biology of melanocytes. Experimental Dermatology [serie en Internet]. Jun 2009 [citado 10 Ene 2012]; 18(9): [aprox. 9 pantallas]. Disponible en: http://onlinelibrary.wiley.com/doi/10.1111/j.1600-0625.2009.00896.x/pdf
2. Matsunaga H, Tanioka M, Utani A, Miyachi Y. Familial case of piebaldism with regression of white Forelock. Clin Exp Dermatol [serie en Internet]. Mar 2008 [citado 10 Ene 2012]; 33(4): [aprox. 2 pantallas]. Disponible en: http://dx.doi.org/10.1111/j.1365-2230.2008.02703.x
3. Haase S, Brooks SA, Tozaki T, Burger D, Poncet PA, Rieder S, et al. Seven novel KIT mutations in horses with white coat colour phenotypes. Animal Genetics [serie en Internet]. May 2009 [citado 10 Ene 2012]; 40(5): [aprox. 7 pantallas]. Disponible en: http://dx.doi.org/10.1111/j.1365-2052.2009.01893.x
4. Piebald Trait PBT [Internet]. OMIM: Johns Hopkins University. [citado 10 Ene 2012]. Disponible en: http://omim.org/entry/172800
5. Leech SN, Moss C. A current and online genodermatosis database. Br J Dermatol [serie en Internet]. Jun 2007 [citado 10 Ene 2012]; 156(6): [aprox. 34 pantallas]. Disponible en: http://dx.doi.org/10.1111/j.1365-2133.2007.07834.x
6. Mollet I, Ongenae K, Naeyaert JM. Origin, clinical presentation and diagnosis of hypomelanotic skin disorders. Dermatol Clin [serie en Internet]. Jul 2007 [citado 10 Ene 2012]; 25(3): [aprox. 9 pantallas]. Disponible en: http://www.sciencedirect.com/science/article/pii/S0733863507000411
7. Perelló Alzamora MR, Alonso San Pablo MT, Unamuno P. Mechón blanco frontal aislado. Diagnóstico y comentario. Piel [serie en Internet]. 2011 [citado 10 Ene 2012]; 26(10): [aprox. 2 pantallas]. Disponible en: http://www.elsevier.es/es/revistas/piel-formacion-continuada-dermatologia-21/mechon-blanco-frontal-aislado-diagnostico-comentario-90039495-casos-diagnostico-soluciones-2011
8. Domínguez Hernández M, León García Y, Fleites Rumbaut M, Lugo Pérez A. Piebaldismo. Presentaciones de casos. Folia Dermatología Cubana [serie en Internet]. 2010 [citado 10 Ene 2012]; 4(1): [aprox. 2 pantallas]. Disponible en: http://www.bvs.sld.cu/revistas/fdc/vol4_1_10/fdc03210.htm
9. Vieira da Silva PC, Rangel P, Couto A. Síndrome de Waardenburg tipo I: relato de caso. Arq Bras Oftalmol [serie en Internet]. 2011 [citado 10 Ene 2012]; 74(3): [aprox. 2 pantallas]. Disponible en: http://www.scielo.br/pdf/abo/v74n3/13.pdf
10. Kozawa M, Kondo H, Tahira T, Hayashi K, Uchio E. Novel mutation in PAX3 gene in Waardenburg syndrome accompanied by unilateral macular degeneration. Eye (Lond) [serie en Internet]. 2009 [citado 10 Ene 2012]; 23 (7): [aprox. 3 pantallas]. Disponible en: http://www.nature.com/eye/journal/v23/n7/pdf/eye2008256a.pdf
Recibido: 24 de febrero de 2012.
Aprobado: 4 de mayo de 2012.
Dra. Deysi Licourt Otero. Especialista de Primer Grado en Medicina General e Integral y Segundo Grado en Genética Clínica. Asistente. Centro Provincial de Genética de Pinar del Río. Correo electrónico: deysili@princesa.pri.sld.cu

{kind=link}
{kind=link}