










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de Ciencias Médicas de Pinar del Río
versión On-line ISSN 1561-3194
Rev Ciencias Médicas vol.21 no.6 Pinar del Río nov.-dic. 2017
PRESENTACIÓN DE CASO
Síndrome de Apert
Apert syndrome
Elsa Camargo Luaces 1, Zulema Serrano Figueroa 2
1 Médica. Especialista Primer Grado Medicina General Integral. Policlínico Universitario Elena Fernández de Castro. Los Palacios. Pinar del Río. Cuba. lirio@Iinfomed.sld.cu
2 Médica. Especialista Primer Grado Medicina General Integral. instructor, Máster en Genética Comunitaria. Policlínico UniversitarioElena Fernández Castro. Los Palacios. Pinar del Río.Cuba. zule70@infomed.sld.cu
Recibido: 22 de mayo de 2017
Aprobado: 2 de septiembre de 2017
RESUMEN
Introducción: el síndrome de Apert es un desorden autosómico dominante,cuyo defecto se produce por una mutación espontánea, que afecta al receptor 2 del factor de crecimiento de los fibroblastos.
Caso clínico: se presentó un caso con síndrome de Apert en una paciente cuya enfermedad le causó retardo en todas las áreas del desarrollo, conocimiento, lenguaje, autoasistencia, social, motora (gruesa y fina) y para lo cual recibió tratamiento médico, quirúrgico y rehabilitador, obteniéndose resultados favorables, proporcionándole una mejor calidad de vida.
Conclusiones: se diagnósticó en la paciente un síndrome dismórficogenético, donde predominó la craneostosis y sindactilia, por lo cual fue valorada por un equipo multidisciplinario, llegándose al diagnóstico de síndrome de Apert.
DeCS: ACROCEFALOSINDACTILIA; CRANEOSINOSTOSIS; SINDACTILIA.
ABSTRACT
Introduction: Apert syndrome is an autosomal dominant disorder, this defect is caused by a spontaneous mutation, which affects receptor 2 of the fibroblast growth factor.
Case report: a case of Apert syndrome was presented in a patient whose disease caused retardation in all areas of development, knowledge, language, self-care, social, motor (gross and fine), she received medical, surgical and rehabilitating treatment, obtaining favorable results, providing a better quality of life.
Conclusion: a genetic dysmorphic syndrome was diagnosed, craniosynostosis and syndactyly predominated; the patient was assessed by a multidisciplinary team, where Apert syndrome was diagnosed.
DeCS: ACROCEPHALOSYNDACTYLIA; CRANIOSYNOSTOSES; SYNDACTYLY.
INTRODUCCIÓN
En 1906, Eugene Apert, un médico francés, describe a nueve pacientes que comparten atributos y características similares, la acrocefalosindactilia;1vocablo cuyo origen deviene de "acro", palabra griega para designar una "cumbre", refiriéndose a la cabeza "puntiaguda" que es característica común en el síndrome, además de "céfalo", que también proveniente del griego significa "cabeza" y finalmentesindactilia que hace referencia a la fusión de dedos de manos y pies. En honor a su descubridor este síndrome es conocido también como Síndrome de Apert.1
El síndrome de Apert es un desorden autosómico dominante cuyo defecto se produce por una mutación espontánea de origen paterno, que afecta al receptor 2 del factor de crecimiento de los fibroblastos (FGFR2).2Aproximadamente dos tercios de los casos son debidos a un cambio de una citosina por una guanina en la posición 755 en el gen FGFR2, lo que causa un cambio de una serina por un triptófano en la proteína.2Se han descrito mutaciones en otros genes como el P253R, relacionado con la sindactilia y el S252W, con el paladar hendido.3
Se calcula una prevalencia de 15 por 1 millón de nacidos vivos, aunque es mayor en los países asiáticos donde puede llegar a 22,3.1, 2
El defecto genético produce un cierre precoz de las suturas craneales, con la subsecuente aparición de craneosinostosis, lo que ocasiona un crecimiento asimétrico de la cabeza, además de una sindactilia simétrica que afecta frecuentemente los cuatro miembros, se acompaña de dismorfia facial y alteraciones neurales, siendo frecuente que aparezca el retardo mental.2 El cerebro normal de un recién nacido necesita espacio para su crecimiento y por eso, normalmente, nace con las suturas abiertas. La caja craneal forma una esfera con piezas en forma de puzle, que no se soldarán hasta varios meses después. Esta semiapertura permite al cerebro, que es de crecimiento muy rápido, adaptarse al cráneo, cuyo ritmo de crecimiento es más lento.1
Cuando una o varias suturas se cierran antes de tiempo, el cerebro se encuentra con una barrera ósea que lo presiona, y al seguir con su proceso natural de crecimiento queda comprimido, produciendo un cráneo inexpansible o craneosinostosis, produciendo una serie de problemas funcionales.2,3
Todos los síndromes acrocefalosindactilares muestran algún nivel de anormalidades en los miembros; sin embargo, las malformaciones típicas que aparecen en las manos de pacientes con síndrome de Apert son distintivas de otros síndromes y siempre muestran cuatro características comunes1:
1. Un pulgar corto con desviación radial.
2. Sindactilia compleja de los dedos índice, mayor y anular.
3. Sinbraquifalangismo.
4. Sindactilia simple en el cuarto espacio interdigital.
PRESENTACIÓN DE CASO
Antecedentes familiares: Nada a señalar
Historia de la enfermedad actual:
Paciente femenina, blanca, de tres años de edad, residente en área rural del municipio Los Palacios.
Madre de 21 años de edad, con antecedentes relativos de salud.
Embarazo no planificado, no deseado, captación tardía, evaluado con riesgo, solamente presentó ingreso en el hogar materno a las 32 semanas por aumento brusco de peso. Parto institucional, distócico por cesárea por presentar líquido amniótico meconial [x], a término, tiempo gestacional 42 semanas y peso 3850 g, talla 50cm, circunferencia cefálica 36cm, circunferencia torácica 38cm, apgar 9-9 puntos, no hipoxia, no íctero. Se mantiene 10 días con ingreso hospitalario por síndrome dismórfico genético con buena evolución y sin complicaciones, el cual estuvo dado por las siguientes características:
Craneosinostosis, fontanela anterior amplia, prominencia frontal con depresión del tercio medio de la cara, órbitas profundas, ojos pequeños, puente nasal plano y ancho, nariz en anteroversión, labios finos, fisura del paladar blando, sindactilia en bloque de los dedos 2,3 y 4 de ambas manos, sindactilia membranosa del dedo 5 de ambas manos, pulgar en autoestopen ambas manos y sindactilia completa en ambos pies, con primer artejo ancho.
Posteriormente es evaluada por un equipo multidisciplinario integrado por Pediatría, Ortopedia, Odontología y Genética Clínica, el cual, basándose en el cuadro clínico presentado por la niña y por estudios complementarios realizados, diagnostica síndrome de Apert.4-6
DISCUSIÓN
El cuadro clínico y examen físico de la paciente corroboran el diagnóstico planteado de síndrome de Apert donde se evidencian las principales características del mismo6 en la paciente:
A nivel craneofacial: craneostosis, disminución de fosas craneales anterior, media y posterior, retardo intelectual, hueso frontal prominente, órbitas poco profundas, hipertelorismo, paladar hendido y puente nasal deprimido (figura 1).
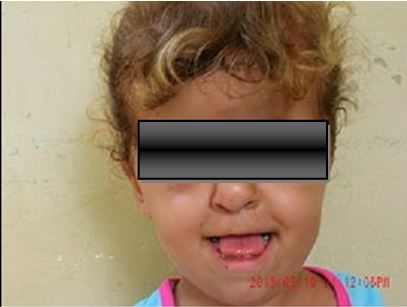
Figura 1
Alteraciones óseas en manos y pies: sindactilia en manos y pies
Tipo I: Incluye el 2do, 3ery 4to dedo.
Tipo II: Asocia el 5todedo.
Tipo III: Todos los dedos aparecen unidos.
En el caso que nos ocupa se trata de un tipo II que asocia el III además (figura 2 y 3)

Figura 2

Figura 3
Alteraciones dermatológicas:Arrugas en frente. Alteraciones estomatológicas: labio superior retruido, forma trapezoidal de la cavidad oral, paladar ojival, fisura palatina, úvula bífida, severo apiñamiento dental, agenesias dentales, erupción retrasada, ectopias dentales, dientes supernumerarios (figura 4).
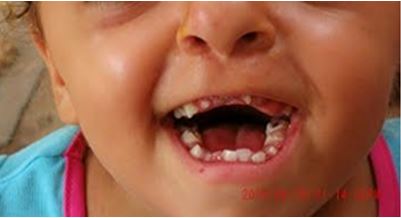
Figura 4
A los dos meses de edad, la paciente es captada en consulta integral de estimulación temprana y después de ser examinadas las diferentes áreas del desarrollo se decide establecer programa de tratamiento rehabilitador individualizado trazando los objetivos según la evaluación y datos positivos en ese momento, empleando kinesiologia para fortalecer la musculatura, mejorar y/o evitar deformidades, terapia ocupacional encaminada a estimular áreas de autoasistencia cognitiva y social y técnicas psicopedagógicas dirigidas a la paciente y la madre para estimular los patrones según áreas del desarrollo y así lograr mejorar su calidad de vida .
La paciente actualmente se mantiene con programa de tratamiento médico rehabilitador por el área de salud, seguimiento en Hospital Pediátrico Juan Manuel Márquez, donde recibió tratamiento quirúrgico para fisuras de paladar y se mantiene en estudio por Ortopedia para posible intervención de la sindactilia.
REFERENCIAS BIBLIOGRÁFICAS
1. Hoyos Serrano Maddelainne, Rojas Mamani Jimmy. SINDROME DE APERT (SA). Rev. Act. Clin. Med [revista en la Internet]. [citado 2017 Jun 13]. Disponible en: http://www.revistasbolivianas.org.bo/scielo.php?script=sci_arttext&pid=S2304-37682014000700008&lng=es.
2. Pérez Breña Ninecta, Abad Aguiar Francisco, Reyes Hernández Dunia, González Martínez Yamil. Síndrome de Apert. Reporte de un caso. MediSur [Internet] 2010 Ago [citado 2017 Jun 13] ; 8( 4 ): 75-77. Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S1727-897X2010000400012&lng=es
3. Reséndiz I. Nava E. Síndrome de Apert. Rev. Acta Médica Grupo Ángeles. [revista en la Internet]. 2013:11 (4). [citado 2017 Jun 13]. Disponible en: http://www.medigraphic.com/pdfs/actmed/am-2013/am134b.pdf
4. Contreras R. Mas F. Jota D. Síndrome de Apert. Reporte de caso en odontopediatría. Odontología Sanmarquina [Internet] 2011. [citado 2017 Jun 13]. 14 (2). Disponible en: http://revistasinvestigacion.unmsm.edu.pe/index.php/odont/article/view/2934
5. Pantoja Ludueña M, Imaña Suarez E, Zeballos Soliz N. Síndrome de Apert. Rev. bol. ped. [Internet]. 2015 [citado 2017 Jun 13] ; 54( 1 ): 24-24. Disponible en: http://www.scielo.org.bo/scielo.php?script=sci_arttext&pid=S1024-06752015000100006.
6. Biblioteca Nacional de Medicina de los Estados Unidos. Síndrome de Apert. Medline Plus. [sitio en Internet]. 2015. [citado 2017 Jun 13]. Disponible en: https://medlineplus.gov/spanish/ency/article/001581.htm
Elsa Camargo Luaces: Médica. Especialista Primer Grado Medicina General Integral. Policlínico Universitario Elena Fernández de Castro. Los Palacios. Pinar del Río. Cuba. Si usted desea contactar con el autor de la investigación hágalo aquí

