










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de Ciencias Médicas de Pinar del Río
versión On-line ISSN 1561-3194
Rev Ciencias Médicas vol.22 no.2 Pinar del Río mar.-abr. 2018
PRESENTACIÓN DE CASO
Poroqueratosis actínica superficial diseminada
Disseminated Superficial Actinic Porokeratosis
Adialys Acosta Rodríguez, 1Javier Martínez Navarro, 2Elizabeth Fernández López 3
1 Médica. Especialista de Primer Grado en Dermatología. Instructora. Hospital General Universitario Dr. Gustavo Aldereguía Lima. Cienfuegos. Cuba. javier.martinez@gal.sld.cu
2 Médico. Especialista de Primer Grado en Anatomía Patológica. Hospital General Universitario Dr. Gustavo Aldereguía Lima. Cienfuegos. Cuba. javiermn@jagua.cfg.sld.cu.
3 Médica. Especialista de Primer Grado en Dermatología. Profesora Asistente. Hospital General Universitario Dr. Gustavo Aldereguía Lima. Cienfuegos.Cuba. elizabeth.fernandez@gal.sld.cu.
Recibido: 11 de diciembre de 2017
Aprobado: 09 de febreo de 2018
RESUMEN
Introducción: la poroqueratosis es un trastorno hereditario de la queratinización. Se describen múltiples variantes clínicas, todas representadas por una lesión primaria común: la pápula hiperqueratósica. El diagnóstico se confirma con la histopatología, donde se aprecia la laminilla cornoide, el hallazgo más representativo en esta dermatosis. La escasa respuesta a la terapéutica, la frecuente recurrencia y su potencial carácter preneoplásico, hacen de esta patología un problema complejo para el dermatólogo. Una de sus variantes; la Poroqueratosis Actínica Superficial Diseminada, es la forma clínica más frecuente con inicio en la tercera y cuarta décadas de la vida, afectando con escaso predominio al sexo femenino.
Objetivo: presentar un caso con poroqueratosis actínica superficial diseminada confirmada por biopsia cutánea en una mujer de 30 años de edad.
Presentación del caso: mujer que acude a consulta de Dermatología presentando lesiones cutáneas acompañadas de prurito intenso localizadas en miembros superiores e inferiores, principalmente en zonas expuestas a la luz solar, de dos años de evolución. En la entrevista médica se constata que la madre presenta lesiones cutáneas similares en zonas fotoexpuestas, cuyo diagnóstico anatomopatológico fue el de poroqueratosis actínica superficial diseminada. El examen físico dermatológico permite sospechar esta entidad, la cual se confirma mediante biopsia.
Conclusiones: a pesar de tratarse de una entidad poco frecuente, el diagnóstico puede realizarse clínicamente teniendo en cuenta la sintomatología, historia familiar, examen físico dermatológico y ser confirmado a través de biopsia cutánea.
DeCS: POROQUERATOSIS; ENFERMEDADES CUTÁNEAS GENÉTICAS; DERMATOLOGÍA.
ABSTRACT
Introduction: porokeratosis is a hereditary disorder of keratinization. Multiple clinical variants are described, all represented by a common primary lesion: the hyperkeratotic papule. The diagnosis is confirmed with histopathological study, where the cornoid lamella is appreciated, the most representative finding in this dermatosis. The poor response to therapy, the frequent recurrence and its potential preneoplastic nature, make this pathology a complex problem for the dermatologists. One of its variants, the disseminated superficial actinic porokeratosis, is the most frequent clinical form with onset in the third and fourth decades of life, affecting with little predominance the female sex.
Objective: to present a case with disseminatedsuperficial actinic porokeratosis(DSAP)confirmed by cutaneous biopsy in a 30-year-old woman.
Case report: a woman who comes to the Dermatology Office presenting skin lesions accompanied by intense pruritus located in the upper and lower limbs, mainly in the areas exposed to sunlight, of 2 years of evolution. In the medical interview, it was found that the mother presented the similar skin lesions in photo-exposed areas, whose pathological diagnosis was disseminated superficial actinic porokeratosis. The dermatological physical examination allows suspecting this entity, which is confirmed by biopsy.
Conclusions: even though this is a rare entity, the diagnosis can be clinically made considering the symptoms, family history, dermatological physical examination, and the confirmation by means of skin biopsy.
DeCS: POROKERATOSIS; GENETIC SKIN DISEASES; DERMATOLOGY.
INTRODUCCIÓN
La poroqueratosis (PQ) es una dermatosis epidérmica que se caracteriza por lesiones anulares, queratósicas e hiperpigmentadas, únicas o múltiples, de evolución crónica. Es una alteración específica de la queratinización y se considera como una lesión premaligna con riesgo elevado de producción de carcinoma epidermoide. Se han reconocido seis variantes clínicas de la enfermedad: la PQ de Mibelli clásica, la PQ superficial diseminada (actínica, no actínica y de inmunosuprimidos), la PQ lineal, PQ palmo-plantar y diseminada y la PQ facial atípica y sindromática. (1, 2) Se considera un trastorno poco frecuente, reportándose a nivel mundial alrededor de 250 casos. (1)
Es considerada una genodermatosis con patrón de herencia autosómico dominante y penetrancia variable, aunque hay casos que resultan de mutaciones espontáneas, así como de múltiples reordenamientos y proliferación anormal de origen clonal de los queratinocitos. Los factores que juegan un papel en la génesis de las lesiones poroqueratósicas son: genético, a la fecha se han demostrado múltiples anomalías genéticas para cada una de las formas clínicas de poroqueratosis y aun es controvertido el aumento en la expresión del gen de factor de supresión p53; exposición a radiación, aunque estudios in vitro sólo han podido demostrar hipersensibilidad de los fibroblastos a la radiación X, pero no a la ultravioleta (UV), en especial en las formas actínicas; traumático, por la presencia del fenómeno isomórfico de Koebner, demostrado en la poroqueratosis clásica de Mibelli y en la lineal; de inmunosupresión, que aumenta en 7.5 a 10 % la frecuencia de poroqueratosis en pacientes postrasplantados o con linfomas, neoplasias hematológicas, infección por virus de inmunodeficiencia humana (VIH) y enfermedades inflamatorias o autoinmunitarias que requieren tratamiento inmunosupresor y, finalmente, enfermedad hepática. (3)
Teniendo en cuenta que se trata de una genodermatosis poco frecuente es que se presenta este caso a la comunidad médica, el cual tuvo diagnóstico anatomopatológico de poroqueratosis actínica superficial diseminada (PASD).
PRESENTACIÓN DEL CASO
Paciente femenina de 30 años de edad, con antecedentes de salud aparente. Acude a consulta de Dermatología presentando lesiones cutáneas acompañadas de prurito intenso localizadas en miembros superiores e inferiores, principalmente en zonas expuestas a la luz solar, de dos años de evolución. Como antecedentes patológicos familiares se constata que la madre presenta lesiones cutáneas similares en zonas fotoexpuestas, lo cual fue confirmado por Anatomía Patológica (AP) mediante biopsia cutánea como una PASD.
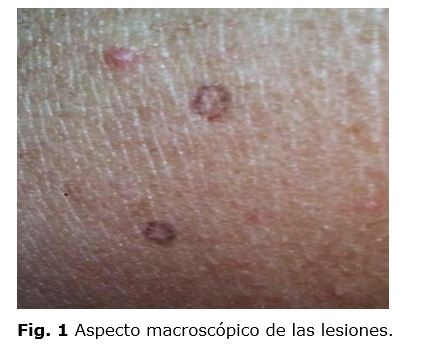
Al examen físico dermatológico se observan placas pequeñas con bordes hiperpigmentados y amurallados localizados en miembros superiores e inferiores. (Fig. 1)
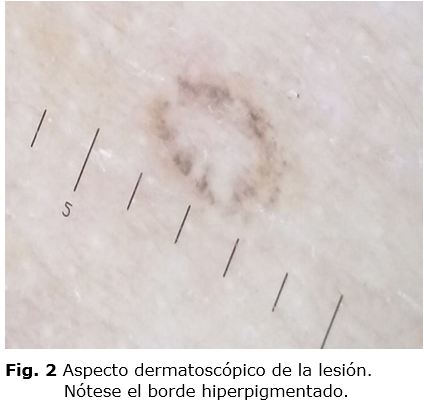
La dermatoscopia permitió ver una lesión que presentó una línea hiperpigmentada y delgada en la periferia en forma de anillo y de color rosado en su centro. (Fig. 2)
Los estudios analíticos realizados fueron normales. Se realiza biopsia cutánea para estudio por AP, donde se informa:
Descripción macroscópica: se recibe fragmento elíptico de piel que mide 2 x 1 x 0.5 centímetros en sus diámetros mayores. Superficie blanquecina con lesión en su centro que muestra borde circular pardo claro.
Diagnóstico microscópico: laminillas cornoides con columna de queratinocitos paraqueratósicos que se extiende desde una invaginación de la epidermis a través del estrato córneo. Atrofia de la capa granulosa subyacente. Ligero infiltrado inflamatorio linfocítico perivascular en dermis superior. Se concluye: PASD. (Fig. 3)

DISCUSIÓN
La PASD es una dermatosis crónica, más generalizada que la poroqueratosis de Mibelli y limitada a las áreas del cuerpo expuestas a la luz solar. Los pacientes refieren exacerbaciones asociadas con prurito y ardor en la piel durante los meses de verano. En el diagnóstico diferencial de esta entidad deben considerarse la acroqueratosis de Hopf, el liquen plano atrófico, la estucoqueratosis, la queratosis actínica, la enfermedad de Bowen, las verrugas planas, la epidermodisplasia verruciforme y la elastosis perforante serpiginosa. (4)
Las lesiones en esta paciente estuvieron localizadas en las extremidades y zonas fotoexpuestas que presentaron prurito intenso.
La PASD, descrita en 1966 por Chernoski y Freeman, es la forma más frecuente de poroqueratosis que comienza hacia la tercera o cuarta década de la vida. Se transmite con un patrón de herencia autosómica dominante, con penetrancia reducida en edades jóvenes y con una incidencia similar en ambos sexos. Se observa con mayor frecuencia en áreas geográficas con mayor grado de exposición solar, y es extremadamente rara en la raza negra. 5En este caso existe el antecedente familiar de madre con lesiones similares en áreas cutáneas fotoexpuestas, en la que también se confirmó mediante biopsia el diagnóstico de PASD.
Las lesiones de PASD son persistentes y no suelen resolverse de forma espontánea, carecen de tratamiento específico; sin embargo, resulta esencial considerar su potencial premaligno. La transformación maligna de las lesiones hacia carcinoma espinocelular y carcinoma basocelular documentada en todas las variantes se estima en 7 %. (3)
El tiempo de evolución de las lesiones cutáneas presentadas por esta paciente fue de dos años.
Los hallazgos dermatoscópicos se correlacionan con sus características histopatológicas. Presenta una fina línea hiperpigmentada en la periferia, y en ocasiones otra línea interna paralela de color blanquecino, que corresponde a la laminilla cornoide. Por dentro de este anillo se pueden observar cordones lineales blanquecinos o amarronados sobre un fondo hiperpigmentado o rosado. También se puede encontrar estructuras vasculares como vasos lineales irregulares o puntiformes. La dermatoscopia no sólo es una técnica diagnóstica en la poroqueratosis, sino también es útil para el seguimiento. Su uso en forma secuencial durante el curso clínico puede mostrar en estadios tempranos el anillo periférico bien demarcado e hiperpigmentado. Mientras en estadios tardíos, donde la atrofia central ya está presente, este anillo se borra en algunos sectores, se hace más difuso y policíclico. (6)Algunas de estas características estuvieron presentes en este caso.
La biopsia debe incluir los bordes y el centro de la lesión para observar los hallazgos característicos de la entidad. Las lesiones típicas suelen diagnosticarse fácilmente, pero deben confirmarse con el estudio histopatológico. El diagnóstico se establece con los cambios observados en la epidermis, en donde se observan columnas de células paraqueratósicas que se distinguen claramente del resto de los corneocitos y que se disponen en «pila de monedas», a lo cual se le conoce como «laminilla cornoide". Por debajo de la columna de células paraqueratósicas, la capa granulosa está ausente o adelgazada. La dermis puede mostrar discretos infiltrados linfocitarios perivasculares. (7, 8)La histopatología es similar en todas las variantes clínicas. (9)Todasestas características estuvieron presentes en el caso que se informa.
Clínicamente se caracteriza por la presencia de pápulas o placas hiperqueratósicas con centro atrófico rodeadas por un borde elevado que histopatológicamente se reconoce como la laminilla cornoide (10) tal y como se manifestó en esta paciente.
REFERENCIAS BIBLIOGRÁFICAS
1. Sánchez Galbán L, Díaz Leonard D, Betancourt Trujillo M. Poroqueratosis. Presentación de un caso. Medisur [Internet]. 2017 [citado 2017 Nov 10]; 15(4): [aprox. 5 p.]. Disponible en: http://www.medisur.sld.cu/index.php/medisur/article/view/3465/2356.
2. Cabrera HN, Mohr YA, Sánchez G. Poroqueratosis genitoglútea: una rara enfermedad habitualmente mal diagnosticada. Dermatol Argent [Internet]. 2015 [citado 2017 Nov 10]; 21 (3): 220-223 [aprox. 3 p.]. Disponible en: http://dermatolarg.org.ar/index.php/dermatolarg/article/viewFile/1423/813
3. Aquino Farrera CJ, López Vázquez F, García Gil A, Toussaint Care S, Lacy Niebla RM. Poroqueratosis superficial diseminada no actínica en una paciente con miocardiopatía dilatada. Dermatol Rev Mex [Internet] 2015[citado 2017 Nov 10]; 59: [aprox. 5 p.]. Disponible en: http://www.medigraphic.com/pdfs/derrevmex/rmd-2015/rmd156j.pdf.
3. Valiente Rebull C, Rodríguez L, Martínez Braga G, Di Martino Ortiz B, Rodríguez Masi M, et al. Poroqueratosis. Reporte de tres casos. Our Dermatol Online [Internet] 2014[citado 2017 Nov 10]; 5(2): [aprox. 5 p.]. Disponible en: http://www.odermatol.com/odermatology/22014/14.Porokeratosis-ValienteRebullC.pdf
4. Bordel MT, Martínez G, Miranda A. Poroqueratosis actínica superficial diseminada asociada a linfedema crónico primario. Actas Dermosifiliogr [Internet] 2004 [citado 2017 Nov 10]; 95(2): [aprox. 3 p.]. Disponible en: http://www.actasdermo.org/es/poroqueratosis-actinica-superficial-diseminada-asociada/articulo/13058578/
5. Pedrozo L, Rodríguez Saa S, Cohen Sabban EN, Cabo H. Dermatoscopía de la poroqueratosis actínica superficial diseminada. Dermatol Argent [Internet] 2015 [citado 2017 Nov 10]; 21 (1): [aprox. 2 p.]. Disponible en: http://www.dermatolarg.org.ar/index.php/dermatolarg/article/viewFile/1390/788.
6. Trejo Acuña JR, Ramos Garibay A, Maza de Franco A. Poroqueratosis actínica superficial diseminada tratada con terapia fotodinámica. Rev Cent Dermatol Pascua [Internet] 2015 [citado 2017 Nov 10]; 24 (1): [aprox. 2 p.]. Disponible en: http://www.medigraphic.com/pdfs/derma/cd-2015/cd151d.pdf
7. Fernández Sánchez C, Galache Osuna C, Hidalgo García Y, Palacio Aller L, Gonzalvo Rodríguez P. Poroqueratosis lineal. Med Cutan Iber Lat Am [Internet] 2015 [citado 2017 Nov 10]; 43 (2): [aprox. 2 p.]. Disponible en: http://www.medigraphic.com/pdfs/cutanea/mc-2015/mc152h.pdf.
8. Saúl LM, Neglia V, Kien MC, Abeldaño A. Poroqueratosis: comunicación de tres casos. Arch Argent Dermatol [Internet] 2016[citado 2017 Nov 10]; 66 (2): [aprox. 2 p.]. Disponible en: http://archivosdermato.org.ar/Uploads/36Saul-Poroqueratosis.pdf.
9. García Vargas Y, Salinas Hojyo RI, Blanco D, Ramírez N. Casuística de Poroqueratosis del Instituto Dermatológico Dominicano y Cirugía de Piel "Dr. Huberto Bogaert Díaz" durante el periodo 2005-2015. Rev Dom Dermatol. [Internet] 2015 [citado 2017 Nov 10]; 42(1): [aprox. 2 p.]. Disponible en: http://revistadominicanadedermatologia.com/wp-content/uploads/2016/07/Volumen_42_No_1_Ene-Jun-2015.pdf.
Adialys Acosta Rodríguez: Médica.Especialista de Primer Grado en Dermatología. Instructora. Hospital General Universitario Dr. Gustavo Aldereguía Lima. Cienfuegos. Cuba. Si usted desea contactar con el autor de la investigación hágalo aquí

