










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Médica Electrónica
versión On-line ISSN 1684-1824
Rev. Med. Electrón. vol.33 no.3 Matanzas mayo-jun. 2011
HOSPITAL PROVINCIAL CLÍNICO QUIRÚRGICO DOCENTE JOSÉ R. LÓPEZ TABRANE. MATANZAS
Klipper Trenaunay Weber. A propósito de un caso
Klipper Trenaunay Weber. A propos of a case
AUTOR
Dr. Ihosvany Ruiz Hernández
Especialista de II Grado en Medicina Interna. Máster en Infectología. Profesor Auxiliar. Hospital Provincial Clínico Quirúrgico Docente José R. López Tabrane. Matanzas.
RESUMEN
Se presentan las características clínicas de un paciente que se concluyó como portador de síndrome Klippel-Trenaunay Weber, quien ingresó en el Hospital Provincial Clínico Quirúrgico Docente José Ramón López Tabrane, de Matanzas, por un hematoma intraparenquimatoso. Este se incluye dentro de los síndromes neurocutáneos con afección vascular. Es de presentación poco frecuente (1/27 500 recién nacidos) y no bien conocido. Posee 3 características fundamentales que lo distinguen: angioma o nevus flammeus, hipertrofia asimétrica de tejidos blandos y várices. En nuestro paciente se observan dichas alteraciones, asociadas a otras anomalías ocasionales.
Palabras clave: clínica, síndrome Klipper Trenaunay Weber, hematoma cerebral, hemangioma cavernoso, hipertrofia, várices.
SUMMARY
We present the clinical characteristics of a patient who was concluded as a carrier of the Klippel-Trenaunay Weber syndrome, who entered the Provincial Hospital of Matanzas José Ramón López Tabranes, with an intraparenchimatous hematoma. It is included among the neurocutaneous syndromes with vascular condition. It is a rare condition (1/27 500 newborns) and it is not well-known. It has three main distinguishing characteristics: angioma or nevus flammeus, asymmetric hypertrophy of soft tissues and varices. Those alterations are observed in our patient, associated to other occasional anomalies.
Key words: clinical, Klipper Trenaunay Weber syndrome, brain hematoma, cavernous hemangioma, hypertrophy, varicose veins.
INTRODUCCIÓN
El Síndrome Klippel-Trenaunay-Weber fue originalmente reportado por Maurice Klippel y P. Trenaunay en 1900. Parkes-Weber F. añadió en 1907 el hallazgo infrecuente de fístula arterio-venosa (1,2). Su etiología no es completamente conocida, constituye una entidad esporádica causada por una mutación en el gen VG5Q involucrado en la angiogénesis y con mosaicismo somático (3), también se ha identificado translocación de 5q y 11p, y anillo supernumerario en el cromosoma 18 que sugieren la posibilidad de mutación única (4). Un patrón de herencia paradominante ha sido planteado a causa de la frecuencia de hemangiomas en varios familiares afectados (5).
Es de presentación poco frecuente (1/27 500 recién nacidos) y se incluye entre los síndromes neurocutáneos, muy numerosos y complejos, los cuales se clasifican respecto a su etiopatogenia en 4 grupos. Uno de ellos incluye aquellos trastornos secundarios a una perturbación en el desarrollo embrionario del Sistema Nervioso y de la piel, los síndromes neurocutáneos con afectación vascular se incluyen en este grupo, sus alteraciones afectan múltiples órganos, pueden consistir en malformaciones de tipo disembriopático o neoplasias (generalmente benignas) (6). El hallazgo clínico más frecuente es el angioma plano o nevus flammeus, las anormalidades pigmentarias son nevus pigmentados y verrugosos en el tipo I, manchas azuladas en el tipo II, nevus spilos en el tipo III, y las dos últimas en el tipo IV. El tipo II es el más frecuente, la mitad de los pacientes tienen compromiso visceral, el Síndrome Klippel-Trenaunay-Weber (SKTW) y la angiomatosis Sturge-Weber (ASW) son las entidades más frecuentes en este grupo (7).
Se diagnostica generalmente en la infancia, por la clínica y las pruebas de imagen (radiografía simple, flebografía, ecografía, TAC o resonancia magnética nuclear), que permiten el diagnóstico prenatal (8-10).
Du y otros, utilizan el término de facomatosis pigmentovascular a la asociación de malformaciones vasculares cutáneas y diferentes desórdenes pigmentarios. La alteración constante de cada tipo es el nevus flammeus. Las anormalidades pig-mentarias asociadas son: nevus pigmentados y verrucoso en el tipo I; manchas azuladas en el tipo II; nevus spilus en el tipo III y manchas azuladas y nevus spilus en el tipo IV. El tipo II es el más frecuente reportado. En este tipo, la mitad de los pacientes tienen compromiso visceral y dentro de él los más habituales son el síndrome de Klippel-Trenaunay-Weber y el síndrome de Sturge-Weber (3).
Causas
La mayoría de los casos de este síndrome ocurre sin una razón aparente; sin embargo, se cree que unos cuantos casos se transmiten de padres a hijos (hereditarios), posiblemente como un rasgo autosómico dominante.
Síntomas
• Muchos hemangiomas planos u otras problemas vasculares, incluyendo manchas oscuras en la piel.
• Venas varicosas (se pueden notar a comienzos de la lactancia, pero se observan con mayor probabilidad posteriormente en la infancia o la adolescencia).
Otros síntomas posibles
• Sangrado del recto
• Sangre en la orina
Pruebas y exámenes
Pronóstico
A pesar de la apariencia estética, la mayoría de los individuos con este síndrome tienen un buen pronóstico; sin embargo, puede haber problemas psicológicos conexos.Nombres alternativos
MÉTODOS
Se realizó una investigación descriptiva, retrospectiva, en el Hospital Provincial Clínico Quirúrgico Docente José R. López Tabrane, de Matanzas, durante el año 2009, sobre un paciente portador de la enfermedad de Klipper Trenaunay Chiari, quien ingresó en la sala J de dicho centro asistencial, comparándose con la revisión bibliográfica referente a esta entidad.
Al paciente se le solicitó consentimiento informado para la presentación de esta investigación, cuidándose los aspectos básicos de la bioética.
Fuentes de información
El dato primario se obtuvo de la revisión de la historia clínica del paciente presentado, así como de la entrevista médica realizada por el autor de la investigación.
Procedimiento
La información relativa a esta investigación fue apoyada por un enfoque médico utilizándose criterios clínicos e imagenológicos en su discusión.
Procesamiento de la información
Los resultados se procesaron en Microsoft Word 2003, empleando una PC Pentium M Celaron 1 300 Mhz, con ambiente de Windows XP.
Síndrome de Klippel-Trenaunay-Weber
Este síndrome consta de:
• Un nevo vascular ubicado en las extremidades inferiores.
• Venas varicosas ubicadas al lado afectado que aparecen al nacimiento o durante la niñez.
• Hipertrofia de los tejidos de la extremidad enferma (principalmente los huesos), posiblemente por hipertensión venosa o éstasis.
La pierna enferma muestra edema y si abarca también el muslo se observa una serie de anormalidades linfáticas como quistes mesentéricos quilosos, quilo peritoneo, y enteropatía perdedora de proteínas. Rara vez se localizan en el aparato genitourinario o gastrointestinal y se manifiesta por hemorragia, la cual puede ser recurrente, leve o abundante y deberse a un hemangioma rectal o vaginal, várices rectovaginales localizadas por obstrucción de las iliacas internas o hipertensión porta con várices. La exploración física es diagnóstica y se utilizan varias técnicas imagenológicas para definir la anatomía y plantear la reparación quirúrgica. Las radiografías simples que muestran flebolitos pélvicos calcificados en niños sugieren hemangiomatosis pélvica.
Sindromografía
Clínica
• Nevo localizado unilateralmente en un miembro.
• Venas varicosas.
• Hipertrofia del tejido blando y huesos del miembro afectado que lo hacen más alargado y caliente que el sano.
Las venas varicosas y la osteohipertrofia no siempre están presentes al nacimiento, pero se desarrollan durante los primeros meses o años de vida. Se asocian frecuentemente al paludismo y sindactilismo.
Exámenes paraclínicos
Radiología: Se observa engrosamiento de la corteza de los huesos, pero eso no constituye un elemento diagnóstico de importancia.
Sindromogénesis y etiología
Posiblemente este síndrome es ocasionado por una debilidad hereditaria del tejido mesenquimatoso de las paredes de los vasos. Ocurre esporádicamente por lo que su etiología no ha podido ser bien precisada.
PRESENTACIÓN DEL CASO
Historia clínica: 750824-05481
Motivo de ingreso: calambres en la mano izquierda.
HEA: Paciente con antecedentes de linfedema crónico y fístula arteriovenosa congénita, para la cual no sigue tratamiento. Acude al cuerpo de guardia por presentar calambres desde hace veinticuatro horas, que comenzaron en la mano izquierda, y se exacerbaban al cambiar de posición la misma, siendo de moderada intensidad y duración variable, desde minutos hasta horas, con una frecuencia de dos o tres episodios al día, de carácter matinal, sin ceder a tratamiento analgésico habitual, posteriormente se corre a la pierna izquierda, con iguales características, acompañándose de dolor de cabeza, desde hace 5 horas, de aparición súbita, el cual es universal, de gran intensidad y no cede a dipirona oral, acompañando este cuadro presentó según sus familiares pérdida del conocimiento que duró varios minutos, orinándose al unísono, sin poder aportar otros datos, por lo que se decide su ingreso para mejor estudio y tratamiento.
Antecedentes patológicos personales: linfedema crónico, fístula arteriovenosa congénita, sarampión, hepatitis viral A, quiste cerebeloso, alergia y linfangitis a repetición.
Hábitos tóxicos: Bebe alcohol frecuentemente. Más de 4 estados de embriaguez alcohólica anualmente.
Examen físico
Peso habitual: 80 kg
Peso actual: 72 kg
Temperatura: 36,6 grados centígrados.
Mucosas: Húmedas y normocoloredas.
Piel y faneras: Telangiectasias en tronco, abdomen y miembro inferior derecho, víbices en cara y planos laterales del tronco.
Respiratorio: Murmullo vesicular disminuido en ambos hemitórax donde se auscultan estertores subcrepitantes aislados. No retracciones intercostales ni subcostales, discreto aleteo nasal, no cianosis. Fr: 24/ rpm. Saturación de oxígeno: 98 %.
Sistema cardiovascular: primer ruido y segundo ruido cardiaco normal, no tercer ruido, ni soplo. Los pulsos periféricos son de buena amplitud e intensidad. El relleno capilar rápido.
Fc: 188/lpm. Tensión Arterial 125/85.
Abdomen: Plano, depresible, no distendido, no visceromegalia, no masas palpables.
Sistema nervioso: Disminución de la fuerza muscular en el hemicuerpo izquierdo, con mayor toma fasciobraquial que crural, disminución de los reflejos osteotendinosos, babinski izquierdo, glasgow 14 puntos.
Exámenes realizados
1. Hb: 13 g/l
2. Leucograma: 8,3 x 10/l
3. Plaquetas: 290x10/l
4. Urea: 3,9 mmol/l
5. Creatinina: 62 mmol/l
6. Proteína C reactiva: 1,6 mg/l
7. Proteínas totales: 74 g/l
8. Albúmina: 27 g/l
9. Bilirrubina total: 22 mmol/l
10. Bilirrubina directa: 6 mmol/l
11. V.S.G: 17 mms
12. Perfil hepático: normal
13. Serologías VIH y VDRL: no reactivas
14. Antígeno de superficie y anticuerpo C: no reactivos
15. A.N.A y ANCA negativos.
16. Lipidograma normal
17. Ultrasonido abdominal normal
18. Rx de tórax: negativo
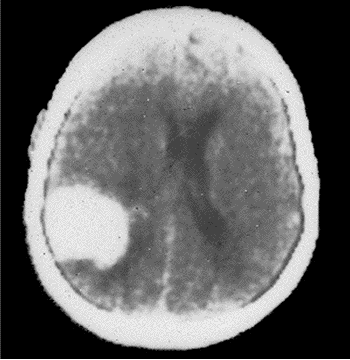
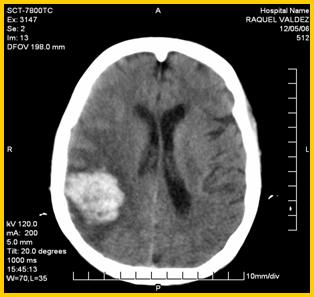
19. TAC de cráneo simple: Se observa lesión hiperdensa parietooccipital derecha. (Figuras 1 y 2)
20. Glicemia: 5,50 mmol.
 |
 |
DISCUSIÓN
El síndrome de Klippel-Trenaunay-Weber (KTW), es un síndrome poco frecuente y no bien conocido entre los médicos, lo que puede conducir a un manejo inadecuado y a un retraso en el diagnóstico de muchos casos incluyendo la demora o no detección de posibles manifestaciones asociadas y que pueden motivar limitación funcional potencialmente evitable en los enfermos (5-9).
Se plantea la posibilidad de relación con el gen para la proteína activadora de la GTPasa, cuya misión es el control del crecimiento y diferenciación celular (2).
La hipertrofia, en la mayoría de los casos, es de las extremidades; puede ser unilateral o afectar una o más de ellas (7,10,11-16).
La hipertrofia es congénita o de aparición temprana en la niñez, usualmente de un miembro 4, el aumento de tamaño puede ser progresivo y afectar a toda la extremidad, a una parte de ella o a los dedos (12-14).
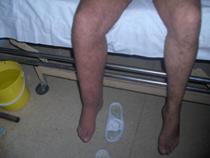
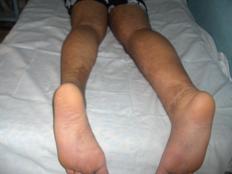
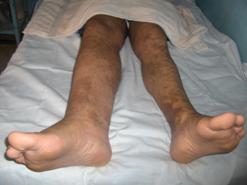
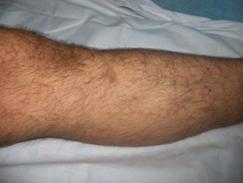
Nuestro paciente presenta hipertrofia de la pierna derecha y en ese nivel presenta un área de hemangioma. (Figuras 3,4,5 y 6)
 |
 |
 |
 |
REFERENCIAS BIBLIOGRÁFICAS
1. Brooksaler F. The angio osteohypertrophy síndrome. Amer J Dis Child. 1996;112:161-4.
2. Klippel M, Trenaunay P. Naevus variqueux osteohypertrophique. Arch Gen Med. 1900;(3):641-2.
3. Weber PF. Angioformation in connection with hypertrophique of limbs and hemihipertrophy. Brit J Derm. 1907;(19):231-5.
4. Garzon M, Huang J, Enjolras O, Frieden I. Vascular malformations. Part II: associated syndromes. J Am Acad Derm. 2007 Apr;56(4):541-64.
5. Redondo P. Clasificación de las anomalías vasculares (tumores y malformaciones). Características clínicas e historia natural. Anales Sis San Navarra. 2004;27(1):9-25.
6. Parcana J, Balaguer M. Síndrome de Klippel-Trenaunay-Weber. Haciendo una unidad clínica. Folia Dermatol. 2004 Sep-Dic;15(3).
7. Tian XL, Kadaba R, YouSA, Liu M, Timur AA, Yang L, et al. Identification of an angiogenic factor that when mutated causes susceptibility to Klippel-Trenaunay syndrome. Nature. 2004; 427:592-4.
8. Timur AA, Sadgehour A, Graf M, Schwart S, Libby ED, Driscoll DJ, et al. Identification and molecular characterization of a "de novo" supernumerary ring chromosome 18 in a patient with Klippel-Trenaunay Syndrome. Ann Hum Genet. 2004;68:353-6.
9. Hofer T, Frank J, Itin PH. Klippel-Trenaunay syndrome in a monozygotic male twin: supportive evidence for the concept of paradominant inheritance. Eur J Dermatol. 2005 Sep-Oct;15(5):341-3.
10. Méndez Sánchez TJ, Otero Alba IC, García García R, Pérez Tamayo B. Síndrome de Klippel-Trenaunay-Weber: presentación de un caso. Rev Cubana Oftalmol. [revista en la Internet ]. 2001 Jun [citado 10 Abr 2009];14(1). Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0864-21762001000100008&lng=es
11. Odom RB, James WB, Berger TG, editors. Andrew's diseases of the skin: Clinical Dermatology. 10 th ed. Philadelphia, PA: Saunders; 2005. p. 585.
12. Avilés Izquierdo JA, Suárez Fernández R, Lázaro Ochaíta P, García Andrade CR. Síndrome de Klippel-Trenàunay. An Med Inter. 2003 Nov;20(11):1-3.
13. Goncalvez LF, Muñoz Rojas MV, Vitorello D, Ternes Pereira E, Pereima M, Saab Neto JA. Klippel-Trenaunay-Weber syndrome pr esenting as massive lymphangiohemangioma of the thigh: prenatal diagnosis. Ultrasound in Obstetrics & Gynecology. 2000,15(6):537-41.
14. Jeong NY, Haeng SK, Hee SR. Prenatal sonographic diagnosis of Klippel-Trenaunay-Weber syndrome: A case report. J Reproductive Med. 2005;50(4):291-4.
15. Garc F. Klippel Trenaunay-Weber Syndrome: A long term study of a singular case" [Abstracts for the 10 th World Congress on Pediatric Dermatology]. Pediatr Dermatol. 2004;21(3):397-8.
16. Lee A, Driscoll D, Gloviczki P. Evaluation and management of pain in patients with Klippel-Trenaunay syndrome: a review. Pediatrics. 2005 Mar;115(3):744-10.
Agradecimientos a: Antonis Cano Soler. Estudiante de cuarto año de la carrera de Medicina y alumno ayudante de Cirugía.
CÓMO CITAR ESTE ARTÍCULO
Ruiz Hernández I. Klipper Trenaunay Weber. A propósito de un caso. Rev Méd Electrón [seriada en línea] 2011;33(3). Disponible en URL: http://www.revmatanzas.sld.cu/revista%20medica/ano%202011/vol3%202011/tema13.htm [consulta: fecha de acceso]

