










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Médica Electrónica
versión On-line ISSN 1684-1824
Rev. Med. Electrón. vol.34 no.2 Matanzas mar.-abr. 2012
PRESENTACIÓN DE CASO
Evolución de paciente pediátrico con Síndrome Alagille. Reporte de caso
Evolution of a pediatric patient with Alagille’s Syndrome. Report of a case
Dra. Estela V. Román Castellini,I Dra. Ibis Umpiérrez García,II Dr. Félix M. Ponce Rodríguez,III Dra. María de los Ángeles López ZayasI
I Hospital Provincial Pediátrico Docente Eliseo Noel Caamaño. Matanzas, Cuba.
II Hospital Militar Docente Mario Muñoz Monroy. Matanzas, Cuba.
III Hospital Clínico Quirúrgico Universitario Comandante Faustino Pérez. Matanzas, Cuba.
RESUMEN
El síndrome de Alagille es una enfermedad congénita y poco frecuente, se transmite de forma autosómica dominante, con expresividad variable. Se caracteriza por presentar colestasis, anomalías vertebrales y oculares, cardiopatía congénita y dismorfias faciales. El pronóstico de este síndrome es variable, depende fundamentalmente de la afectación hepática y los defectos cardiovasculares asociados. Se presentó el caso de una paciente con diagnóstico de síndrome de Alagille con evolución estable.
Palabras clave: afectación hepática, autosómica dominante, defectos cardiovasculares, enfermedad congénita, niño, síndrome de Alagille.
ABSTRACT
The Alagille’s syndrome is a few frequent congenital disease; it is transmitted in a dominant autosomal way, with variable expressivity. It characterizes for presenting cholestasis, vertebral and ocular anomalies, congenital cardiopathies and facial dysmorphias. The prognosis of this syndrome is variable, mainly depending of the hepatic injury and the associated cardiovascular defects. We presented the case of a patient with the diagnosis of Alagille’s Syndrome and stable evolution.
Key words: hepatic damage, autosomal dominant, cardiovascular defects, congenital disease, child, Alagille’s Syndrome.
INTRODUCCIÓN
El síndrome de Alagille es una enfermedad congénita y poco frecuente, se transmite de forma autosómica dominante, con expresividad y fenotipo variable,(1) se caracteriza por la presencia de hipoplasia de vías biliares intrahepaticas asociado a un cuadro de colestasis intrahepática crónica,(2) debido a la escasez de los conductos hepáticos que generalmente se inicia en el período neonatal, se asocian además anomalías cardíacas, oculares, esqueléticas, y la presencia de un fenotipo peculiar que según algunos autores es típico y diagnóstico. Alteraciones menos frecuentes son las anomalías glomerulares, alteraciones de la voz, genitales, dentales, oído interno o en la capacidad intelectual dado por retraso mental moderado.(3)
La afectación hepática se presenta desde la etapa neonatal o lactante pequeño con colestasis en el 70 % de los casos y 17 % tienen manifestaciones cardiovasculares.
El diagnóstico suele hacerse generalmente en el primer año de vida, aunque se han reportado casos de diagnósticos más tardíos.(4)
La prevalencia de este síndrome es de 1/70 000 niños nacidos vivos en España;(2) Estados Unidos, 1 por 100 000 niños nacidos;(4) y en Cuba, Castañeda y cols. han presentado varios casos.(5)
Se reporta el presente caso con el objetivo de describir el cuadro clínico y la evolución de una paciente pediátrica con diagnóstico de Síndrome de Alagille en la primera infancia, la cual se mantiene en seguimiento.
PRESENTACIÓN DEL CASO
Paciente de 8 años de edad, sexo femenino, antecedentes maternos de dos abortos espontáneos anteriores, producto de embarazo con amenaza de aborto, por lo cual se mantuvo en reposo, nacida de parto eutócico a término (40 semanas), con Apgar 9/9 y peso de 2 760 gramos.
Al mes de nacida presentó un cuadro de ictericia y coluria que alterna con orinas normales, es valorada en consulta detectándose un soplo cardíaco, por lo que se ingresa para estudio.
Examen físico
Mucosas húmedas con cianosis peribucal.
Piel: Ligero tinte subictérico y cianosis distal.
Aparato respiratorio: Polipnea, no tiraje, murmullo vesicular audible no estertores.
Frecuencia respiratoria: 52xss
Aparato cardiovascular: Ruidos cardíacos taquicárdicos: 1er ruido normal, 2do ruido aumentado y único, 3er ruido y retumbo en ápex, se ausculta soplo sistólico lll/Vl en 3er espacio intercostal izquierdo.
Frecuencia cardíaca: 142xss
Abdomen: Globuloso, suave, depresible, no doloroso a la palpación, hepatomegalia de 1,5 cm, ruidos hidroaéreos presentes.
Sistema nervioso central: Fontanela normotensa, reflejos presentes y normales para su edad.
Resto del examen físico sin alteración aparente.
Exámenes complementarios
Gasometría: pH 7,33, pCO2 44,8 mmHg, pO2 39,3 mmHg SO2 70.3 %
Bilirrubina total: 6,7 mg%, Bilirrubina directa: 6,1 mg%, bilirrubina indirecta: 0,6 mg% Hemoglobina 14,9 gr/l, Hematocrito: 0,48, Eritro: 2 mm, Plaquetas 211x109/l, Transaminasa glutámico pirúvica: 38UI, Transaminasa glutámico oxalacética: 29 UI
Radiografía de tórax: Aumento del índice cardiotorácico biventricular, aumento del flujo pulmonar, no lesiones inflamatorias.
Ecocardiograma: Situs solitus, ápex izquierdo concordancia veno-atrial y atrio ventricular, salida única arterial tipo tronco, crecimiento biventricular, crecimiento auricular izquierdo, comunicación interventricular de bordes musculares de 8 mm tronco conal sobre el cual cabalga el único vaso concluyéndose como tronco común.
Ultrasonido abdominal: Hígado de 1 cm, de ecoestructura normal, vesícula, bazo, riñones normales.
Se inicia tratamiento con digoxina, furosemida, cloruro de potasio, mejorando el cuadro, siendo dada de alta con seguimiento diez días después del ingreso.
Durante el primer año de vida presentó varios ingresos por signos de descompensación cardiovascular que mejoró con tratamiento. En su seguimiento se le realizaron nuevos estudios concluyéndose como cardiopatía congénita compleja (atresia pulmonar más comunicación interventricular) continuando con tratamiento y seguimiento.
Posteriormente, a los 2 años de edad, el tinte ictérico se intensifica, aumentando el cuadro de colestasis, el cual se hizo progresivo, se reevalúa la paciente detectándose al examen físico:
Mucosas: húmedas, tinte ictérico y cianosis peribucal.
Piel: Xantomas cutáneos planos en los codos y prurito.
Uñas en vidrio de reloj, dedos en palillo de tambor.
Aparato respiratorio: Polipnea ligera, no tiraje, murmullo vesicular audible no estertores. Frecuencia Respiratoria: 42xss
Aparato cardiovascular: Ruidos cardíacos taquicardicos: 1er ruido normal, 2do ruido aumentado y único, soplo sistólico lll/Vl en 3er espacio intercostal izquierdo. Frecuencia cardíaca: 122xss
Abdomen: suave, depresible, no doloroso a la palpación, hepatomegalia 2 cm, no esplenomegalia, ruidos hidroaéreos presentes.
Resto del examen físico sin alteración.
Se realizan exámenes complementarios con los siguientes resultados: (Véase tabla)
Fosfatasa alcalina: 2120 u/l, Transaminasa glutámico pirúvica: 92 UI, Transaminasa glutámico oxalacética: 29 UI, Proteínas Totales: 58g/l, Albúmina: 25 g/l, Bilirrubina total: 2,0 mg%, Bilirrubina directa: 1,8 mg%, Bilirrubina indirecta: 0,2 mg%, Hb 16,8 g/L, Hto 0,58, Eritro: 4 mm/h, Ecografía abdominal: hígado 2 cm vesícula, bazo y riñones normales, estudios virológicos para citomegalovirus y herpes virus con resultados negativos, continuando tratamiento y seguimiento de su enfermedad de base.
Comienza a los 3 años con aumento de volumen de las rodillas y dolor a la movilización de las mismas con persistencia de la colestasis y aumento del prurito, se ingreso para estudio.
Examen físico
Cara: Frente prominente, hipertelorismo, nariz bulbosa.
Mucosas: Ictéricas, húmedas, y cianosis peribucal.
Piel: Ligero tinte ictérico, xantomas cutáneos planos en codos y aumento del prurito.
Uñas en vidrio de reloj, dedos en palillo de tambor.
Aparato respiratorio: Polipnea ligera, no tiraje, murmullo vesicular audible no estertores. Frecuencia Respiratoria: 34xss.
Aparato cardiovascular: Ruidos cardíacos rítmicos: 1er ruido normal, 2do ruido único, se ausculta soplo sistólico lll/Vl en 3er espacio intercostal izquierdo. Frecuencia Cardiaca: 106xs
Abdomen: suave, depresible, no dolor a la palpación, hepatomegalia 1cm, no esplenomegalia, ruidos hidroaéreos presentes.
SOMA: Aumento de volumen de ambas rodillas y tobillo izquierdo, no calor, no rubor, no impotencia funcional, ligera atrofia de ambos cuádriceps con deformidad de ambas rodillas.
Se indican estudios virológicos: Antígeno superficie hepatitis virus B no reactivo, anticuerpo hepatitis virus C no reactivo, IgM hepatitis virus A no reactivo, citomegalovirus y herpes virus negativo.
Ecografía abdominal: Hígado de ecoestructura homogénea, tamaño normal, vesícula de tamaño normal con paredes ligeramente engrosadas, bazo de tamaño y ecoestructura normal, páncreas normal.
Se reevalúa la paciente con estos elementos clínicos tenemos tres criterios mayores planteándose como diagnóstico Síndrome de Alagille basados en lo siguiente:
1. Colestasis crónica con debut en el período neonatal.
2. Malformación cardiovascular: atresia pulmonar + comunicación interventricular (aunque lo más frecuente es las estenosis de la arteria pulmonar).
3. Fascie característica: Frente prominente, hipertelorismo, nariz bulbosa, mentón afilado.
Se indican otros estudios complementarios (tabla):
Radiografía de columna total: No se observan vertebras en mariposa.
Examen oftalmológico con lámpara de hendidura: No se encontró embriotoxón posterior.
Resultados de los principales complementarios
| Complementarios | Resultados |
| Hemoglobina | 15,9 (g/l) |
| Velocidad sedimentación globular | 9 (mm/l) |
| Leucograma | 10,7 (x109/l) |
| Neutrófilos | 58 (%) |
| Linfocitos | 34 (%) |
| Monocitos | 5 (%) |
| Eosinófilos | 3 (%) |
| Conteo de plaquetas | 220 (x109/l) |
| Tiempo de coagulación | 2 |
| Tiempo de sangramiento | 6 |
| Transaminasa glutámico pirúvica | 80 UI |
| Transaminasa glutámico oxalacética | 53 UI |
| Fosfatasa alcalina | 2574 u/l |
| Gammaglutamil transpeptidasa | 928 u/l |
| Bilirrubina Total | 6,5 mgr% |
| Bilirrubina Directa | 6,0 mgr% |
| Bilirrubina Indirecta | 0,5 mgr% |
| Proteínas totales | 78 gr/l |
| Albúmina | 34 gr/l |
| Glicemia | 4,2 mmol/l |
| Colesterol | 7,9 mmol/l |
| Triglicéridos | 4,9 mmol/l |
| Creatinina | 51 mmol/l |
| Calcio sérico | 8,6 mg% |
| Fósforo | 1,0 mmol/l |
Fuente: historia clínica
Por todo lo anterior, además del tratamiento de base, se inicia tratamiento con Ibuprofeno a 50 mg/kg/día para la sintomatología articular y de sostén, para la colestasis con Calcio 600 mg/día, vitamina D 200 UI/día, vitamina E 50 mg/día, vitamina A y D2, 20 gotas/día cada 10 días, aceite oliva 5 cc 2v/día, sulfato de zinc 5 cc 2 v/día, acido ursodeoxicólico ¼ tableta diaria y fenobarbital 50 mg/día.
Se da seguimiento a la paciente por el cuadro articular que evolucionó como una artritis franca (pauciarticular), con buena respuesta al tratamiento antinflamatorio, con diagnóstico de oligoartritis seronegativa en el curso del síndrome de Alagille. (Fig. 3)
Posteriormente, continuó su evolución estable con tratamiento durante el cuarto y quinto año de vida; se valoró la realización de biopsia hepática, cuyo resultado anatomopatológico de cilindro hepático de morfología en general conservada donde se observan dos espacios porta y uno de sus extremos con cambios grasos macro vacuolares.
En su seguimiento posterior hasta la actualidad, la paciente se ha mantenido con una evolución estable, se han realizaron exámenes complementarios con parámetros aceptables en su seguimiento.(Véanse figs. 1 y 2)
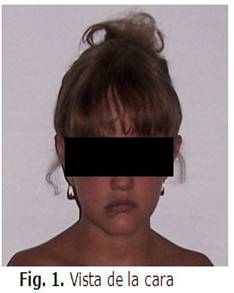
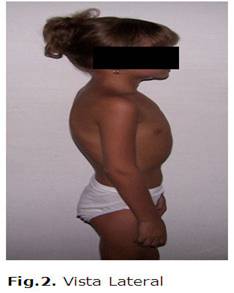
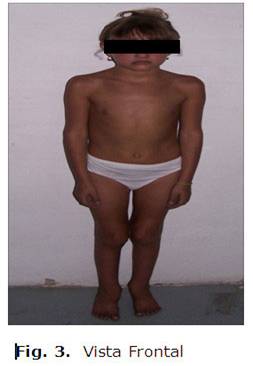
DISCUSIÓN
El Síndrome de Alagille es un trastorno genético hereditario de carácter autosómico dominante y expresión variable, con compromiso del cromosoma 20p12: donde se encuentra el gen JAG I, que codifica un ligando del receptor transmembrana Notch, estando involucrado en los procesos de diferenciación celular y morfogénesis que afecta estructuras dependientes del mesodermo durante el desarrollo embrionario.(6)
Entidad poco frecuente, que cursa con colestasis crónica neonatal o tardía, entre el 45 a 75 % de los casos presentan ictericia temprana, presentándose en el 100 % de los pacientes a los 2 años de vida.(7) Las manifestaciones aparecen entre los 2 y 3 meses de edad, relacionadas con la colestasis, realizándose el diagnóstico en los primeros meses de vida en la mayoría de los casos. En el caso que se presenta se realizó el diagnóstico a los 36 meses, a diferencia de lo reportado por Alagille y cols.(3) en su investigación, coincidiendo con Ruiz, donde en su estudio reportaron casos que se diagnosticaron hasta los 74 meses de vida,(8) pues algunos niños no tienen todas las alteraciones que describen este síndrome, ya que las alteraciones cromosómicas que provocan esta enfermedad, ocurren en el 88 % de los casos con mutaciones en el gen JAG 1, mientras el 7 % de los casos presentan algunas delecciones.
La tríada característica diagnóstica en esta enfermedad que más se describe es la colestasis, estenosis pulmonar, la facies peculiar, en este caso la paciente presentó atresia pulmonar,(9) colestasis y la fascie, otras manifestaciones clínicas asociadas fueron detención de peso y talla, prurito intenso secundario a la colestasis por depósito de pigmentos biliares en el tejido celular subcutáneo, el cual se presenta generalmente después del sexto mes de vida al igual que lo reportado por otros autores.(10) Actualmente, se plantea el diagnóstico con la presencia de cuatro o más criterios mayores en los casos en que no se demostró paucidad ductal en la biopsia hepática, o en los que esta no se realizó.(8)
La alteración histológica es progresiva, no es habitual verla en la etapa de recién nacido, aunque esta se considera la manifestación más importante del síndrome, puede no estar presente en todos los afectados.
El pronóstico a largo plazo depende de la severidad del daño hepático y las malformaciones cardiovasculares asociadas. Entre el 21 % y 31 % de los pacientes requieren trasplante hepático,(11) por el desarrollo de cirrosis con insuficiencia hepática, hipertensión portal, prurito incoercible, mientras un 15 % puede desarrollar complicaciones como hepatocarcinoma, insuficiencia hepática, insuficiencia pancreática exocrina, fibrosis hepática con hipertensión portal.
La mortalidad es aproximadamente del 10 %, debido a fallo cardíaco, fallo hepático severo.
El manejo multidisciplinario, con un tratamiento encaminado a mejorar el funcionamiento del corazón, reducir los efectos causados por el deterioro de la función hepática, manejo de la colestasis,(12) uso de citoprotectores, lo cual ha demostrado que disminuye las cifras de ALT y los niveles de colesterol, mejorando el prurito, soporte nutricional que proporcione un adecuado aporte de energía, suplementación de vitaminas liposolubles (A, D, E, K), mejorando el estado nutricional, lo que permitirá establecer diagnósticos oportunos e impactar de manera positiva en la calidad de vida del paciente.
REFERENCIAS BIBLIOGRÁFICAS
1. Alagille D, Odièvre M, Gautier M, Dommergues JP. Hepatic ductular hypoplasia associated with characteristic facies, vertebral malformations, retarded physical, mental, and sexual development murmur. J Pediatr [Internet]. 1975 [citado 14 Dic 2011];86:63-71. Disponible en: http://www.jpeds.com/article/S0022-3476%2875%2980706-2/abstract
2. Frauca RE, de la Vega A. Colestasis en el lactante. En: Jara P, editora. Trasplante hepático en niños. Madrid: ERGON; 2006. p. 23-35.
3. Alagille D, Estrada A, Hadchouel M, Gautier M, Odievre M, Dommergues JP. Syndromic paucity of interlobular bile ductus (Alagille syndrome or arterihepatic dysplasia): review of 80 cases. J Pediatr. 1987;110(2):195-200. PubMed; PMID: 3806290.
4. Fadlouallah M, Krami H, Tamzaourte M, Lahmiri M, Tahri S, El Koundi H, et al. Alagille syndrome diagnosed in adolescence: a case report. Arch Pediatr [Internet]. 2010 Jul [citado 12 Dic 2011];17(7):1111-3. Disponible en: http://preview.ncbi.nlm.nih.gov/pubmed?term=Alagille%20syndrome%20diagnosed%20in%20adolescence%3A%20a%20case%20report
5. Castañeda C, Fragoso Trini, GB, Guerra L, Castellanos O, Trujillo ME. El síndrome de Alagille en Cuba: informe de 9 casos. GEN. 1992 [citado 12 dic 2011];46(4): 341-6. PubMed; PMID: 1340842.
6. Crosnier C, Attie-Bitach T, Encha-Razavi F, Audollent S, Soudy F, Hadchouel M, et al. Jagged1 gene expression during human embryogenesis elucidates the wide phenotypic spectrum of Alagille syndrome. Hepatology. 2000;32:574-81. PubMed; PMID: 10960452.
7. Jiménez Jiménez JR, Castellanos Reyes K, Huerta Albarrán R, Edith Justiniani Cedeño N, Yáñez López MP, Sierra Tortolero A. Un caso del síndrome de Alagille. Rev Mex Pediatr [Internet]. 2007 [citado 21 Dic 2011];74(4):152-7. Disponible en: http://www.medigraphic.com/pdfs/pediat/sp-2007/sp074d.pdf
8. Ruiz Castillo MA, Michel Peñichel F, Cervantes Bustamante R, Zárate Mondragón F, Mata Rivera N, Montijo Barrios E, et al. Síndrome de Alagille: informe de 12 casos en el Instituto Nacional de Pediatría. Rev Enf Infec Pediatr [Internet]. 2007 [citado 21 Dic 2011]; 81:13-7. Disponible en: http://www.medigraphic.com/pdfs/revenfinfped/eip-2007/eip073d.pdf
9. Lemione TJ, Kaza AK, Gray R, Day RW, Tani LY, Poss W. Anomalous origin of the left coronary artery from the pulmonary artery in Alagille syndrome: Alagille Síndrome. Congenit Heart Dis. 2010 Sep-Oct;5(5):462-4. PubMed; PMID: 21087434.
10. Arocena E, Machado K, Pirez MC, Montano A. Síndrome de Alagille. A propósito de un caso. Arch Pediatr Urug [Internet]. 2010 [citado 21 Dic 2011];81(3):158-162. Disponible en: http://www.scielo.edu.uy/scielo.php?pid=S0004-05842010000300004&script=sci_arttext
11. Mata Zubillaga D, Iglesias Blázquez C, Herrero Mendoza B, Rodríguez Fernández C, Lapeña López de Armentía S. Síndrome de Alagille y trasplante hepático: Caso clínico. Bol Pediatr [Internet]. 2008 [citado 21 Dic 2011];48:276-8. Disponible en: http://www.sccalp.org/documents/0000/0160/BolPediatr2008_48_276-278.pdf
12. Zellos A, Roy A, Schwarz KB. Use of oral naltrexone for severe pruritus due to cholestatic liver disease in children. J Pediatr Gastroenterol Nutr [Internet]. 2010 Dec [citado 21 Dic 2011]; 51(6):787-9. Disponible en: http://journals.lww.com/jpgn/Citation/2010/12000/Use_of_Oral_Naltrexone_for_Severe_Pruritus_Due_to.18.aspx
Recibido:18 de enero de 2012.
Aprobado:15 de febrero de 2012.
Estela V. Román Castellini. Hospital Provincial Pediátrico Docente Eliseo Noel Caamaño. Matanzas. Matanzas, Cuba. Correo electrónico: vilmar.mtz@infomed.sld.cu
CÓMO CITAR ESTE ARTÍCULO
Román Castellini EV, Umpiérrez García I, Ponce Rodríguez FM, López Zayas MA. Evolución de paciente pediátrico con Síndrome Alagille. Reporte de caso. Rev Méd Electrón [Internet]. 2012 Mar-Abr [citado: fecha de acceso];34(2). Disponible en: http://www.revmatanzas.sld.cu/revista%20medica/ano%202012/vol2%202012/tema12.htm
