










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Médica Electrónica
versión On-line ISSN 1684-1824
Rev.Med.Electrón. vol.39 no.3 Matanzas may.-jun. 2017
PRESENTACIÓN DE CASOS
Epidermólisis bullosa: piel de mariposa. A propósito de un caso
Epidermolysis bullosa: butterfly skin. Apropos of a case
MSc. Odalis de la Caridad del Rosario Marrero,I MSc. Yordanka Smith Ordoñez,II Est. Adrián Luis González Jústiz,I Est. Ailin González Díaz,I Est. Aylet Arcis del Rosario,I MSc. Yumila Fernández LeónI
I Filial Universitaria de Ciencias Médicas. Dr. Eusebio Hernández Pérez. Matanzas, Cuba.
II Hospital General Docente “Dr. Mario Muñoz Monroy”. Matanzas, Cuba.
RESUMEN
La epidermólisis bullosa comprende un grupo heterogéneo de enfermedades ampollosas de la piel y las mucosas, son de origen congénito y hereditario. Hacer el diagnóstico no es difícil si se tiene experiencia dermatológica, pero su clasificación es compleja y para ello se necesita considerar la clínica, la genética, la microscopia y la evaluación de laboratorio. El tratamiento de esta enfermedad es también dificultoso y son necesarias ciertas medidas, para proteger al paciente, evitar la aparición de lesiones y las complicaciones derivadas de ellas. Se describe el tratamiento de estas lesiones en un recién nacido, al que se administraron antibióticos profilácticos y se colocaron vendajes en las lesiones. Se describieron todos los cuidados y recomendaciones para evitar, especialmente los roces y las presiones en estas lesiones, así como las temperaturas altas. Para la confección del presente trabajo se consultaron 18 materiales entre revistas y libros de Pediatría. El caso reportado fue un recién nacido con epidermólisis bullosa atendido en el Hospital Universitario “Dr. Mario Muñoz Monroy” de Colón, Matanzas. Se demostró lo poco frecuente y raro de esta patología para los especialistas del tema.
Palabras clave: epidermólisis bullosa, recién nacido, lesiones.
ABSTRACT
The epidermolysis bullosa includes a heterogeneous group of bullous skin and mucous diseases of congenital and hereditary origin. Diagnosing them is not difficult if the specialist has dermatologic experience, but their classification is complex and it is necessary to take into account the clinical, genetic and microscopic factors, and the laboratory assessment. The treatment of this disease is also difficult and it is necessary to take certain measures to protect the patient, avoid the onset of lesions and the complications derived from them. The treatment of these lesions in a newborn is described. Prophylactic antibiotics were administered and bandages were put on the lesions. All the cares and recommendations to avoid rubbings and pressures on these lesions, and also the high temperatures, are described. To develop the current term, 18 materials (journals and pediatric books) were consulted. The reported case was the case of a newborn with epidermolysis bullosa attended in the University Hospital “Dr. Mario Muñoz Monroy” of Colon, Matanzas. It was demonstrated the low frequency and rarity of this pathology for the specialists of the theme.
Key words: epidermolysis bullosa, newborn, injuries.
INTRODUCCIÓN
La epidermólisis bullosa (EB) o ampollar es un conjunto de enfermedades o trastornos de la piel transmitidas genéticamente. Se manifiestan por la aparición de ampollas, úlceras y heridas en la piel, en especial en las áreas mucosas al más mínimo roce o golpe, también suelen aparecer heridas internas, provocando un cierre en el esófago lo que provoca pérdida de peso al no poder digerir alimentos, pero el problema de esófago, estómago y heridas internas, generalmente ocurre en la epidermólisis bullosa distrófica recesiva.
Suele manifestarse al nacer o en los primeros meses de vida, y existen dos formas en las que la enfermedad se puede heredar.(1)
1. En la herencia dominante, uno de los progenitores tiene la enfermedad y existe 50 % de probabilidades por cada embarazo, que nazca un bebé afectado.
2. En la herencia recesiva, ambos progenitores (padres) son portadores de un gen enfermo que trasmite la enfermedad. En cada embarazo existe el riesgo de 1 a 4 (25 %) de que tengan un bebé enfermo con EB.
La epidermólisis bullosa congénita es una enfermedad que se observa rara vez, ya que su prevalencia es del orden de 32 casos por millón de habitantes, con una incidencia de casos nuevos de 1,4 por millón de habitantes por año, y con variaciones que dependen más de la buena calidad de los registros, que de diferencias regionales o étnicas. Por esta razón es un desafío médico pasar del diagnóstico sindrómico al específico, con el fin de orientar a los padres sobre la naturaleza, pronóstico de la enfermedad, y establecer un manejo actualizado tanto para el período agudo como para la etapa ambulatoria posterior, y, por último, advertir sobre la posibilidad de ocurrencia en gestaciones posteriores.(1,2)
La piel de los afectados con epidermólisis bullosa se caracteriza por ser frágil, débil, extremadamente sensible y muy vulnerable, tan delicada como el cristal; ya que al menor contacto físico se les desprende la piel, causándoles heridas y ampollas que tienen el aspecto de una gran quemadura. Es por esto que la enfermedad también recibe el nombre de enfermedad piel de cristal o piel de mariposa.(3)
Hacer el diagnóstico no es difícil si se tiene experiencia dermatológica, lo que resulta complicado es su clasificación, y para ello se necesita considerar la clínica, genética, microscopia y evaluación de laboratorio.(4)
En cuanto a la EB distrófica recesiva, quienes presentan las formas más graves pueden llegar a la muerte de manera directa por la enfermedad o indirectamente por sus complicaciones. En las formas menos graves, el pronóstico es relativamente bueno, aunque estas personas estarán debilitadas durante toda su vida.(5)
En el hospital de Colón se presentó un caso clínico de un recién nacido en el que se precisó que la enfermedad ampollar estaba localizada a nivel subepidérmico. Resulta de interés presentar dicho caso y hacer una revisión del tema desde una perspectiva multidisciplinaria.
PRESENTACIÓN DEL CASO
Paciente recién nacido en el hospital “Dr. Mario Muñoz Monroy”, del Municipio de Colón, Matanzas, el 9 de mayo de 2008. Hija de una mujer de 24 años de edad, raza blanca, G1 P0 A0 con edad gestacional de 41,4 semanas, con antecedentes personales de sepsis urinaria y sepsis vaginal, que fueron tratadas con ampicillin y metronidazol respectivamente.
El bebé nace producto de un parto por cesárea primitiva debido a una desproporción cefalopélvica, con un tiempo de rotura de membranas de dos horas.
Desde el momento del nacimiento se detectan lesiones ampollares denudadas, profundas en los dos pies, con atrofia de uñas, lesiones ampollares con contenido amarillento claro, localizadas en zonas de roce, labios, mucosa bucal y lengua, así como una fragilidad cutánea generalizada que se evidencia cuando se le aplica la inyección de vitamina K para la profilaxis de la enfermedad hemorrágica, lo que trajo como consecuencia la pérdida de la epidermis. (Fig. 1,2 y 3)
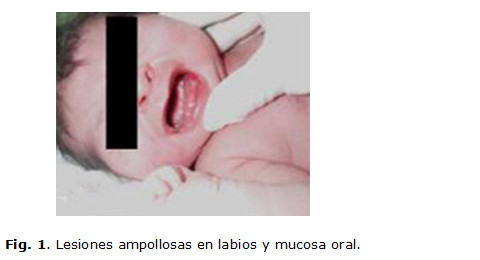
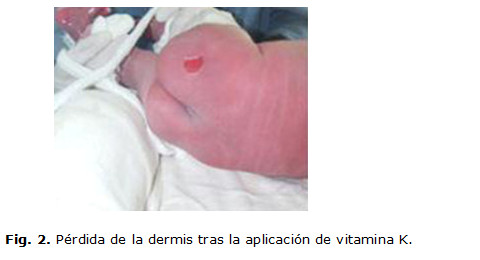
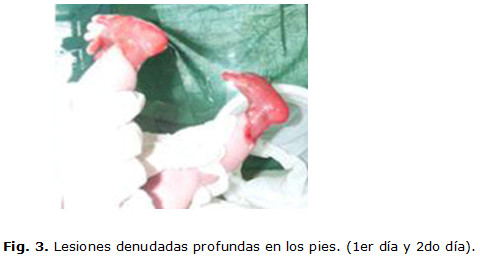
Se le realizaron complementarios como hemograma, glicemia, proteína C con valores normales. Progresivamente aparecieron nuevas lesiones ampollares en las mejillas, las manos, el abdomen y los muslos. (Fig. 4 y 5)
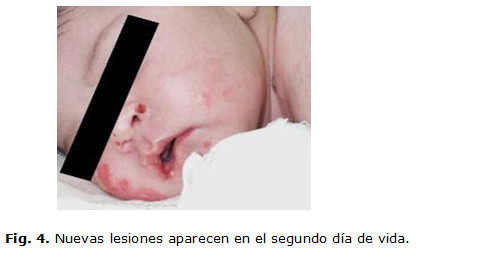
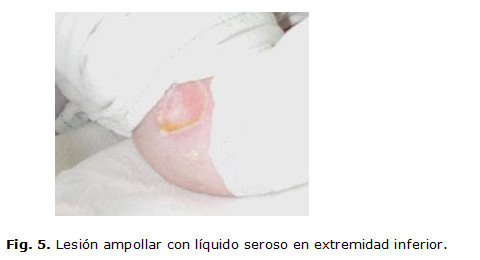
Fue valorada por Dermatología se le indicó tratamiento antibiótico oral, tópico profiláctico y vendajes húmedos oclusivos. Evolutivamente aparecieron nuevas lesiones con contenido hemorrágico y ocurrió la epitelización de otras lesiones que dejaron cicatriz.
La lactancia materna favoreció la mejoría de las lesiones bucales. Fue dada de alta porque mantuvo una favorable evolución, al transcurrir un mes y medio aproximadamente acuden al centro por presentar numerosas lesiones y se decidió su ingreso. Se mantuvo bajo supervisión y cuidados médicos intensivos durante once meses y unos días, cumpliendo su primer año de vida hospitalizada. Posteriormente fue dada de alta por poseer en su hogar las condiciones necesarias, como una ventilación adecuada de su habitación para mejor evolución de su enfermedad.
DISCUSIÓN
En la actualidad, el diagnóstico específico se hace comúnmente por estudio histopatológico con microscopia electrónica de transmisión, complementado con microscopia convencional, análisis histoquímico y estudio molecular de genes de las proteínas involucradas. La microscopia electrónica permite clasificar este síndrome congénito en tres grupos principales, de acuerdo al plano de separación en que se produce la ampolla, estos son:(6)
Simple: es causada por una mutación de las células basales de la epidermis que se manifiesta en lesiones en manos y pies. La rotura se produce en la capa superficial de la piel (epidermis), las ampollas cicatrizan sin pérdida de tejido y los afectados suelen experimentar mejoría con el tiempo. Este tipo de EB se manifiesta en un 52,5 % de los casos.
Juntural: es producida por una mutación de la proteína laminina 5 que une las membranas y puede afectar las mucosas oculares, cavidad oral, vía urinaria, esófago y faringe. Las ampollas aparecen en la zona situada entre la capa externa y la interna, los subtipos que incluyen van desde una variedad letal hasta otros que pueden mejorar con el tiempo. Existen muy pocos casos diagnosticados con esta variedad (1 % de los casos).
Distrófica: las ampollas aparecen en el estrato más profundo de la piel, la dermis. Al cicatrizar, las sucesivas heridas van originando retracciones en las articulaciones, llegando a dificultar seriamente el movimiento (las heridas pegan la piel de entre los dedos). También pueden aparecer ampollas en las membranas mucosas de: boca, faringe, estómago, intestino, vías respiratorias y urinarias e interior de los párpados y córnea. Este tipo de EB se manifiesta en un 46,5 % de los casos.(1-7)
En los tipos de la unión y distrófico se han descrito alteraciones en algunas de las ultraestructuras que forman la lámina lúcida o densa de la membrana basal, respectivamente. Para estos casos es muy útil la técnica inmuno histoquímica, con anticuerpos monoclonales contra distintos subtipos de colágeno, en particular IV y VII. Esto permite identificar si el colágeno está en el piso o en el techo de la ampolla subepidérmica; así se pueder clasificar la lesión en sus variantes principales, simple y distrófica.(7,8)
El estudio ultraestructural permite definir el tipo y variante de epidermólisis bullosa en la mayoría de los casos, pero debe complementarse con la información obtenida por las otras técnicas, porque así se minimiza la posibilidad de un error en la interpretación de los hallazgos morfológicos.(9,10) En el caso que se estudió se realizaron técnicas descritas anteriormente y el diagnostico se realizó por observación clínica.
La facilidad de infección de las lesiones dificulta el diagnóstico morfológico, complica el manejo, retarda la cicatrización, prolonga la hospitalización y es causa frecuente de septicemias. En la paciente estudiada, las ampollas presentaban inicialmente un trasudado claro, como profilaxis se efectuó un tratamiento antibiótico orientado contra cocáceas grampositivas y se protegió las lesiones con gentamicina. La rápida regresión del exudado y la ausencia de necrosis perilesional no hicieron necesario considerar el uso de otros antibióticos.(9)
Los hemocultivos negativos, el hemograma y PCR normales no apuntaron a infección sistémica. En este síndrome es difícil decidir cuándo iniciar una cura antibiótica basándose sólo en criterios clínicos, y más controvertidos aun, es su suspensión. La ausencia de rodete inflamatorio y la falta de progresión de las lesiones, una vez vaciadas y comprimidas, son consideraciones locales que orientan hacia la ausencia de infección.(10)
La mayor preocupación de los padres y doctores que atienden a estos pacientes son las infecciones, ya que estas pueden llegar a la sangre y de ahí pueden pasar al corazón u otro órgano. No obstante, el cuidado adecuado y meticuloso puede hacer que el paciente tenga una buena inmunidad, pero no hay que confiarse; a primera vista es como una persona quemada en forma crónica y superficial, lo que es muy doloroso e invalidante, porque los enfermos deben ser sometidos diariamente a largas curaciones, así lo explica Sprecher E,(11) en sus estudios.
Los dermatólogos consideran que el tratamiento consiste principalmente en un manejo adecuado de la enfermedad. Se realizan curaciones con vendas, mallas de vaselina y cremas antibióticas para contrarrestar las infecciones. También se les protege con vendas especiales y se advierte de no usar telas adhesivas porque se quedan con la piel al retirarlas, los recién nacidos deben usar la ropa al revés y esta debe ser 100 % de algodón, hay que puncionar las ampollas y evitar las infecciones.(11)
Para las manos y pies recogidos se realizan cirugías reconstructivas que devuelven estas partes a su forma original. Cuando el compromiso esofágico llega a una estenosis-estrechez es necesario efectuar cirugía.(12)
Actualmente se administra hierro intravenoso ya que las personas que padecen esta enfermedad pueden presentar problemas de cicatrización puesto que desarrollan anemia. Además, se suministra un suplemento de vitamina D, puesto que estos niños no pueden sintetizar esta vitamina debido a que su piel, al estar cubierta por vendas, no tiene contacto con el sol. Para el problema del cierre de esófago suelen hacerse operaciones para que puedan alimentarse bien.(9)
El periodo de hospitalización, en este caso, permitió educar a los padres en aspectos claves del cuidado de la niña en el hogar y de asegurar el apego a los controles. Se les explicó la necesidad de evitar los ambientes calurosos y a preferir ropas de algodón, se les adiestró en el uso de vendajes que cubran manos y pies, así como la necesidad de cuidados extremos en la colocación y retiro de estas vendas, ya que además del riesgo de lesionar la lámina ungueal; se pueden producir ampollas interdigitales de difícil cicatrización y que tienden a generar pseudomembranas con fusión de los dedos, si no se les venda en forma individual. La necesidad de proteger la piel más vulnerable a los traumatismos, como codos y rodillas, fue otra de las cuestiones explicadas a los padres. Todas estas medidas son recomendadas por la literatura consultada.(2,9-12)
La infección sobreaguda de las lesiones, habitualmente por Staphylococcus aureus, con dolor local y fiebre, es una emergencia que puede producir una septicemia fulminante si no se toma la precaución de hospitalizar al paciente para un tratamiento especializado.(13)
Los aspectos nutricionales merecen una consideración especial, pues la reparación casi constante de las bulas produce un elevado consumo calórico-proteico, en un paciente que habitualmente se alimenta mal, tanto por las frecuentes lesiones del tubo digestivo, como por las carencias específicas producto de la alternancia subinstante de lesiones y cicatrizaciones. Por esta razón, en los pacientes con vesículas orales se aconseja mantener la alimentación con papillas, cuidando de aportar las vitaminas, oligoelementos y hierro en dosis mayores a los requerimientos.(13-15)
Pese a ello, se ha visto que las formas más invalidantes presentan un raquitismo larvado, agravado por la inmovilización.(15,16)
Se considera que otra consecuencia del acelerado recambio epitelial, es la posibilidad de degeneración maligna con aparición de carcinomas escamosos en piel o en la mucosa digestiva, a partir de la segunda década de la vida.(17)
La epidermólisis bullosa distrófica recesiva después de la EB de unión tipo Herlitz es la variante más grave de trastorno mecanoampolloso y origina mutilación, debilidad y muerte temprana en muchos casos. Esta variante incluye un rango de gravedad clínica muy voluble.(18)
REFERENCIAS BIBLIOGRÁFICAS
1- McKenna K, Walsh M, Bingham E. Epidermolysis bullosa in Northern Ireland. Br j dermatol [Internet]. 1992 [citado 15 Sep 2015];127(4):318-21. Disponible en: http://onlinelibrary.wiley.com/doi/10.1111/j.1365-2133.1992.tb00448.x/full
2- Horn H, Tidman M. The clinical spectrum of dystrophic epidermolysis bullosa. Br j Dermatol [Internet]. 2002 [citado 15 Sep 2015];146(2):267-74. Disponible en: http://onlinelibrary.wiley.com/doi/10.1046/j.1365-2133.2002.04607.x/pdf
3- Chung Jin H, Uitto J. Epidermolysis bullosa with pyloric atresia. Dermatologic clinics [Internet]. 2010 [citado 28 May 2015];28(1):43-54. Disponible en: http://www.sciencedirect.com/science/article/pii/S0733863509000795
4- Sánchez Macias LR, García Retana PP, Viego Romero ME. Epidermólisis bullosa congénita (piel de cristal). Acta méd centro [Internet]. 2012 [citado 28 May 2015];6(4). Disponible en: http://www.medigraphic.com/pdfs/medicadelcentro/mec-2012/mec124n.pdf
5- Gupta R, Woodley DT, Chen M. Epidermolysis bullosa acquisita. Clinics in dermatology [Internet]. 2012 [citado 28 May 2015];30(1):60-9. Disponible en: http://www.sciencedirect.com/science/article/pii/S0738081X11000873
6- Intong LR, Murrell DF. Inherited epidermolysis bullosa: new diagnostic criteria and classification. Clinics in dermatology [Internet]. 2012 [citado 28 May 2015];30(1):70-7. Disponible en: http://www.sciencedirect.com/science/article/pii/S0738081X11000885
7- Fine JD, Eady RA, Bauer EA, et al. Revised classification system for inherited epidermolysis bullosa: Report of the Second International Consensus Meeting on diagnosis and classification of epidermolysis bullosa. AAD [Internet]. 2000 [citado 15 Sep 2015];42(6):1051-66. Disponible en: http://scholar.google.com.cu/scholar?q=Revised+classification+system+for+inherited+epidermolysis+bullosa%3A+report+of+
the+second+international+consenses+meeting+on+diagnosis+and+classification+of+epidermolysis+bullosa&btnG=&hl=es&as_sdt=0%2C5#
8- Itoh M, Kiuru M, Cairo MS, et al. Generation of keratinocytes from normal and recessive dystrophic epidermolysis bullosa-induced pluripotent stem cells. Proceedings of the National Academy of Sciences [Internet]. 2011 [citado 28 May 2015];108(21):8797-802. Disponible en: http://www.pnas.org/content/108/21/8797.short
9- Pope E, Lara-Corrales I, Mellerio J, et al. A consensus approach to wound care in epidermolysis bullosa. JAAD [Internet]. 2012 [citado 28 May 2015];67(5):904-17. Disponible en: http://www.sciencedirect.com/science/article/pii/S0190962212001016
10- Sawamura D, Nakano H, Matsuzaki Y. Overview of epidermolysis bullosa. The Journal of dermatology [Internet]. 2010 [citado 28 May 2015];37(3):214-9. Disponible en: http://onlinelibrary.wiley.com/doi/10.1111/j.1346-8138.2009.00800.x/full
11- Sprecher E. Epidermolysis bullosa simplex. Dermatologic clinics [Internet]. 2010 [citado 28 May 2015];28(1):23-32. Disponible en: http://www.sciencedirect.com/science/article/pii/S0733863509000771
12- Schober Flores C. Epidermolysis bullosa: a nursing perspective. Dermatol nurs [Internet]. 1999 [citado 15 Sep 2015];11(4):243-8, 53-6. Disponible en: http://europepmc.org/abstract/med/10670355
13- Vargas A, Palomer L, Palisson F. Manifestaciones orales de la epidermólisis bulosa en el niño. Rev chil pediatr [Internet]. 2005 [citado 15 Sep 2015];76(6):612-6. Disponible en: http://www.scielo.cl/scielo.php?script=sci_arttext&pid=S0370-41062005000600009
14- Smith LT. Ultrastructural findings in epidermolysis bullosa. Arch Dermatol [Internet]. 1993 [citado 15 Sep 2015];129(12):1578-84. Disponible en: http://archderm.jamanetwork.com/article.aspx?articleid=555415
15- Hovnanian A, Christiano AM, Vitto J. The molecular genetics of dystrophic epidermolysis bullosa. Archi Dermatol [Internet]. 1993 [citado 15 Sep 2015];129(12):1566-70. Disponible en: http://archderm.jamanetwork.com/article.aspx?articleid=555397
16- Tolar J, Xia L, Riddle MJ, et al. Induced pluripotent stem cells from individuals with recessive dystrophic epidermolysis bullosa. Journal of Investigative Dermatology [Internet]. 2011 [citado 28 May 2015];131(4):848-56. Disponible en: http://www.sciencedirect.com/science/article/pii/S0022202X15352301
17- Uitto J, McGrath JA, Rodeck U, et al. Progress in epidermolysis bullosa research: toward treatment and cure. Journal of Investigative Dermatology [Internet]. 2010 [citado 28 May 2015];130(7):1778-84. Disponible en:
http://encedirect.com/science/article/pii/S0022202X15348818
18- Wagner JE, Ishida Yamamoto A, McGrath JA, et al. Bone marrow transplantation for recessive dystrophic epidermolysis bullosa. New England Journal of Medicine [Internet]. 2010 [citado 28 May 2015];363(7):629-39. Disponible en: http://www.nejm.org/doi/full/10.1056/NEJMoa0910501
Recibido: 17 de septiembre de 2015.
Aceptado: 14 de marzo de 2016.
Odalis de la Caridad del Rosario Marrero. Filial Universitaria de Ciencias Médicas. “Dr Eusebio Hernández Pérez”. Maceo No. 261, e/ López Coloma y Narciso López. Colón. Matanzas, Cuba. Correo electrónico: omarrero.mtz@infomed.sld.cu

