










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Médica Electrónica
versión On-line ISSN 1684-1824
Rev.Med.Electrón. vol.39 no.3 Matanzas may.-jun. 2017
PRESENTACIÓN DE CASOS
Angiomatosis encefalotrigeminal o síndrome de Sturge-Weber. A propósito de un caso
Encephalotrigeminal angiomatosis or Sturge-Weber syndrome. A propos of a case
Dra. Meylín de la Caridad Morales Querol,I Est. Enrique Marcos Sierra Benítez,II Est. Mairiannys Quianella León Pérez,II Est. José Antonio Márquez Oquendo,II Est. Lisbet Robles Lorenzo,II Lic. Tamay Caridad López ÁlvarezIII
I Hospital Territorial Docente Dr. Mario Muñoz Monroy. Matanzas, Cuba.
II Filial Universitaria de Ciencias Médicas Dr. Eusebio Hernández Pérez. Matanzas, Cuba.
III Policlínico Comunitario Francisco Figueroa Veliz. Matanzas, Cuba.
RESUMEN
El síndrome de Sturge-Weber consiste, en su forma completa, en la asociación de anomalías cerebrales, cutáneas y oculares. Desde el punto de vista clínico, se caracteriza por una mancha color vino en la cara, epilepsia, retraso mental, otras manifestaciones neurológicas deficitarias y glaucoma. Se presentó el caso de un paciente de 9 años de edad, masculino, piel negra, producto de parto eutócico, con peso de 4 200 g y antecedentes patológicos perinatales de cianosis, constatándose desde el nacimiento varios hemangiomas faciales. Al examen físico se confirma en cara y cráneo, una frente huidiza con presencia del hemangioma facial o hemangioma coroideo, glaucoma unilateral izquierdo, micrognatia. Se precisó mala oclusión dentaria, y en las extremidades superiores una clinodactilia del quinto dedo. Además, tiene el tronco asimétrico, retraso mental severo y aparecen cuadros convulsivos tónico-clónicos que repiten frecuentemente. Teniendo en cuenta todos estos elementos se comienza a sospechar el posible diagnóstico de un síndrome de Sturge-Weber.
Palabras clave: síndrome de Sturge-Weber, angioma.
ABSTRACT
The Sturge-Weber syndrome is, in its complete form, the association of ocular, cutaneous and cerebral anomalies. From the clinical point of view, it is characterized by a wine-color spot on the face, epilepsy, mental retardation, other neurological deficit manifestations and glaucoma. We presented the case of a male, black patient, aged 9 years, who was born by eutocic delivery, with 4 200 g weight, and perinatal symptoms of cyanosis, finding several facial hemangioma from the moment of birth. At the physical examination, it was confirmed on the face and crania, evasive forehead with facial hemangioma or choroidal hemangioma, left unilateral glaucoma, micrognathia. Dental malocclusion was stated and clinodactyly of the fifth finger. Besides that, he has an asymmetric trunk, severe mental retardation and tonic-clonic convulsive episodes frequently repeated. Taking into account all these elements we began to suspect the possible diagnosis of a Sturge-Weber syndrome.
Key words: Sturge-Weber syndrome, angioma.
INTRODUCCIÓN
La angiomatosis encefalotrigeminal descrito por Sturge(1) en 1879 y perfeccionada por Weber(2) en 1922, es un proceso congénito infrecuente, pero no excepcional, que afecta a los dos sexos por igual,(3-6) aunque se han descrito casos heredados de forma autosómica recesiva y dominante.(3,7) El síndrome de Sturge-Weber (SSW) consiste, en su forma completa, en la asociación de anomalías cerebrales (angioma leptomeníngeo o pial), cutáneas (angioma facial) y oculares (angioma coroideo). Desde el punto de vista clínico, se caracteriza por una mancha color vino en la cara, epilepsia, retraso mental, otras manifestaciones neurológicas deficitarias (hemiparesia, hemianopsia) y glaucoma. Las formas incompletas del síndrome aparecen como:(3,7-9)
-Angioma facial y leptomeníngeo, pero sin angioma coroideo.
-Angioma leptomeníngeo y coroideo sin nevus facial.
-Nevus facial y angioma coroideo sin evidencias clínicas ni radiográficas de angiomatosis cerebral.
-Angiomatosis cerebral y pial aislado.
-Angioma cutáneo mucoso.
La incidencia de angioma plano facial en la población es de 0,3 %,(4) pero solo alrededor del 10 % tiene una lesión neurocular, y el diagnóstico de SSW debe ser reservado para estos individuos. Es un nevus vascular plano y homogéneo, raramente tuberoso, de color rojo vino, que afecta la cara, y se asienta sobre el párpado y la región frontal ipsilateral al angioma pial. Con frecuencia, el angioma afecta a la hemicara completa, y puede extenderse al cuello, tronco y extremidades de forma uni o bilateral.(5,7,10) Los miembros inferiores se afectarán más que los superiores. El nevus puede involucrar la mucosa de los labios, paladar, lengua, mejilla, faringe y laringe; en estos casos puede haber sangramiento.(3) Las partes blandas de la cara pueden estar hipertrofiadas, con menos frecuencia; partes duras, subyacentes (huesos, dientes).(6) Generalmente el angioma es unilateral, pero entre el 145 y el 30 %(3,10) según la serie, puede ser bilateral. La extensión extrafacial es más frecuente cuando el angioma es bilateral (53 %), que cuando es unilateral (10 %). Un aspecto en la definición del SSW, sujeto a controversia, es lo restrictivo de los criterios que algunos de los autores expresan en cuanto a la topografía del angioma. Así, para algunos, el angioma puede distribuirse por cualquiera de los 3 territorios de las ramas sensitivas del trigémino; para otros, solo si el angioma afecta a los territorios de las ramas oftálmicas (VI) o maxilar (V2) del nervio hay posibilidad de afectación neurocular.(5,6,9) El nevusfacial está presente desde el nacimiento y no cambia de aspecto con la edad,(3) y cuando el niño no tiene evidencia de afectación neurológica, ofrece serias dificultades el determinar si desarrollará o no disfunción neurológica, pues algunos niños con SSW son neurológicamente normales por varios meses o años después del parto.(4)
Afectación ocular
El angioma ocular aparece en el 30 % de los casos,(7) y afecta a la coroides y la esclerótica ocular, y es ipsilateral al angioma cutáneo. La exploración del fondo de ojo muestra una elevación anaranjada localizada en el polo posterior del ojo. Este angioma coroide produce glaucoma entre el 25 y el 60 % de los casos, según la serie,(3,4,10) siempre ipsilateral al angioma facial cuando este afecta el territorio de las ramas oftálmicas (VI) y maxilar (V2) del trigémino.(5) El glaucoma puede ser unilateral o bilateral (siempre que las lesiones cutáneas sean bilaterales), aparece en los primeros años de la vida o puede ser congénito, casos en el que puede producir buftalmo.(3,4) Los niños que muestran angiomas faciales extensos(4) o de localización extrafacial(5) presentan glaucoma con mayor frecuencia. Clínicamente se manifiesta por dolor retroorbitario y deterioro de la visión. En algunos casos puede observarse heterocromía del iris, angioma orbitario y dilatación de los vasos retinianos.
Manifestaciones neurológicas
La afectación neurológica del SSW se relaciona con la presencia de un angioma leptomeníngeo que afecta la piamadre. Este angioma pial puede aparecer aislado, sin angioma de las coroides ni facial, en el 13 % de los enfermos.(7,8,11) Lo más constante es la presencia de crisis epilépticas, que afectan entre el 75 y el 90 % de los pacientes.(3,8-10) En el 45 % de estos las crisis se inician antes del año, con un pico entre los 3 y 6 meses.(3) Inicialmente predominan las crisis parciales motoras o tonicoclónicas generalizadas. Los espasmos infantiles, las crisis mioclónicas y atónicas ocurren con menos frecuencia.(4,7)
Aproximadamente el 7 % de los pacientes inicia sus crisis después de los 5 años.(3) Es usual la presencia de crisis frecuentes y prolongadas de difícil control farmacológico. El inicio de las crisis por debajo de los 2 años hace más probable su refractariedad y el subsecuente retraso mental.(4) El control total de las crisis se logra en el 47 % de los enfermos;(8) otros refieren que las crisis epilépticas son más frecuentes cuando existe afectación bihemisférica.(4) El retraso mental afecta entre el 50 y el 70 %(4) de los pacientes con SSW, y su presencia se hace notar con posterioridad al inicio de las crisis epilépticas. Su intensidad, relacionada con el comienzo precoz de las crisis y su refractariedad,(4,10) puede variar desde ligera hasta profunda, y es más intensa en los pacientes con lesiones corticales bilaterales, en los cuales el 8 % de estos son intelectualmente normales.(4) El 2,5 % de los enfermos desarrolla retraso mental severo.(8-10) Los signos de déficit neurológico focal que se muestran con mayor frecuencia son: defecto motor hemiparético, hemianopsia y hemiatrofia corporal, y, en general, se observan en el 65 % de los enfermos. El defecto hemiparético que se aprecia en el 30 al 40 % de los casos coincide con el comienzo de las crisis epilépticas,(3,4) y muchas veces se atribuye erróneamente al estado poscrítico (parálisis de Todd); sin embargo, persiste durante un tiempo mayor que pocas horas o puede hacerse permanente. Posteriormente, los pacientes pueden presentar episodios de hemiparesia sin crisis previas, que agravan la hemiparesia preexistente.(3) En el 31 % de los pacientes existe hemiatrofia de los miembros contralaterales al angioma pial.(7)
Excepcionalmente, en casos de angiomatosis cutánea y pial bilateral, puede observarse un defecto tetraparético.(3) En un grupo de pacientes se muestran defectos del campo visual, que en dependencia de la localización del angioma pueden presentarse como hemianopsia o cuadrantanopsia homónimas e incluso ceguera cortical cuando existe lesión bilateral de la corteza occipital.(3,4)
PRESENTACIÓN DEL CASO
Paciente de 9 años de edad, masculino, de piel negra, producto de parto eutócico con peso de 4 200 g y antecedentes patológicos perinatales de cianosis, constatándose desde el nacimiento varios hemangiomas faciales. En los primeros meses de vida, en su seguimiento por genética y neurología, al examen físico se confirma una frente huidiza con presencia del hemangioma facial en cara y cráneo (figuras 1 y 2) y la presencia de un hemangioma coroideo, (figura 3) glaucoma unilateral izquierdo, micrognatia. También se precisa mala oclusión dentaria, y en las extremidades superiores, una clinodactilia del quinto dedo.
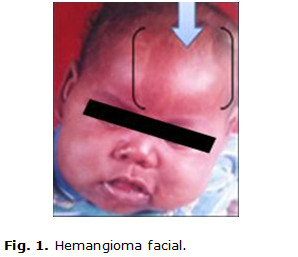
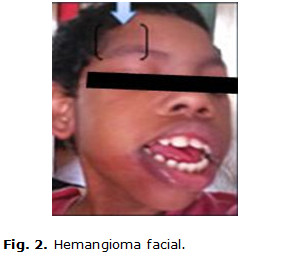
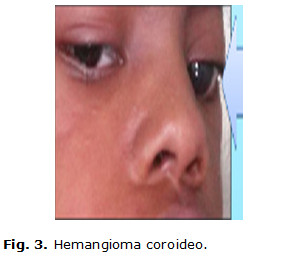
Además, posee tronco asimétrico, retraso mental severo y aparecen cuadros convulsivos tónico clónicos, que repiten frecuentemente y a veces hasta 40 veces en el día. Las convulsiones eran de muy difícil control, y le indican tratamiento con piracetan, vitamina B6 (piridoxina), carbamazepina y clobazán. Teniendo en cuenta todos estos elementos, se comienza a sospechar el posible diagnóstico de un Síndrome de Sturge-Weber.(11)
Examen complementario
Resonancia magnética nuclear (RMN) de cráneo (figura 4). Corte coronal en T2: arrojó una atrofia cortical del hemisferio derecho con desplazamiento de la línea media.(11)

DISCUSIÓN
Debido a que muchos niños con angioma intracraneal son inicialmente normales, los estudios de neuroimagen y otros complementarios pueden ayudarnos a distinguir los que pertenecen al SSW. La marca radiográfica de esta enfermedad constituyen las calcificaciones corticales giriformes adyacentes al angioma leptomeníngeo. Pueden observarse mediante rayos X o tomografía axial computadorizada (TAC) de cráneo, y se encuentran generalmente en la región occipital, aunque es común su presencia en otras áreas.(3,4)
La presencia de angioma pial puede demostrarse con TAC, pero las calcificaciones pueden enmascararlo. La RMN sin contraste muestra imágenes de atrofia y, en algunos casos, calcificaciones como áreas de vacío de señal. La RMN con administración endovenosa de gadolinio es el medio más fidedigno para valorar la extensión del angioma pial,(9,10) que aparece como una imagen que llena el espacio subaracnoideo, y cubre la superficie de los surcos.(8)
La atrofia cerebral en el área del angioma puede distinguirse mediante TAC; sin embargo, la RMN puede mostrar cambios atróficos más sutiles. La materia blanca subcortical se observa hipodensa en la TAC y con alta señal en T1 y T2, debido a la gliosis secundaria por la isquemia cerebral.(10) La hemiatrofia cerebral puede provocar asimetría craneal, el diploe óseo más grueso y los senos perinasales más grandes en el lado afectado. Las estructuras de la línea media pueden verse desplazadas hacia el lado afectado. Los plexos coroides ipsilaterales a la lesión están frecuentemente hiperplasiados, lo cual parece relacionado con una malformación angiomatosa.(6,9)
Los estudios de imagenología estructural (TAC y RMN) son de utilidad para evaluar:(5)
-Extensión de la malformación angiomatosa leptomeníngea.
-Grado de atrofia del parénquima.
-Posibles cambios isquémicos que afectan sustancia gris y blanca.
-Presencia y extensión de calcificaciones corticales.
-Prominencia de plexos coroides.
-Anomalías venosas del parénquima.
-Prominencia diploica del lado afectado.
Varios estudios demuestran la mayor eficacia de la RMN craneal para la evaluación de la afectación neurológica del SSW, sobre todo cuando se utiliza contraste paramagnético.(5,8,9)
La exploración electroencefalográfica puede ayudar, inicialmente, a confirmar la presencia de afectación cerebral y, más tarde, a localizar el sitio de inicio de las crisis epilépticas. Los trazados intercríticos de vigilia muestran una asimetría de la actividad de base, con caída de voltaje en el área del angioma.(3,4) La actividad paroxística puede ser focal, multifocal o generalizada, simétrica o asimétrica, y se origina, generalmente, en el hemisferio ipsilateral al angioma y aumentar durante la hiperventilación.(7,10) En algunos pacientes con angioma pial unilateral pueden observarse paroxismos generalizados y bisincrónicos.(9) Los estudios de metabolismo cerebral mediante tomografía de emisión de positrones han mostrado una reducción del metabolismo cerebral cerca del angioma que se extiende bien por toda el área radiográfica anormal.(8) La tomografía de emisión de fotón simple y el estudio de flujo sanguíneo cerebral usando inhalación de xenón-133, demuestran la reducción de la perfusión en el tejido cerebral afectado.(7-10) Los estudios de imagenología funcional complementan a las estructurales para la evaluación del tratamiento quirúrgico en estos pacientes.
Es un síndrome de etiopatogenia desconocida, aunque parece deberse a un deficiente desarrollo de la vascularización embriológica, por un error que afecta específicamente a una zona de la cresta neural, y que es la responsable del origen el tejido conectivo de la dermis facial, la coroides ocular y la piamadre. El síndrome de Sturge-Weber no tiene un patrón genético claro y no existe evidencia directa de predisposición hereditaria. No existen reportes de mujeres que hayan tenido más de un hijo con este síndrome, más aún, casi nunca aparecen dos individuos afectados en una misma familia. El síndrome se presenta en todas las razas y con igual frecuencia en ambos sexos.(7-10)
Aunque la causa exacta del síndrome de Sturge-Weber es desconocida, en algunos casos se cree que pueda ser un desorden hereditario autonómico dominante. Los rasgos humanos, incluyendo las enfermedades genéticas clásicas, son el producto de la interacción de dos genes, uno recibido del padre y otro de la madre. En los desordenes dominantes, una copia única del gen enfermo (recibido del padre o la madre) se expresará como dominante sobre el otro gen normal y resultará en la apariencia de la enfermedad. El riesgo de transmitir el desorden del padre afectado a su descendencia es de 50 % en cada embarazo, independientemente del sexo resultante en el niño.(7)
Las leptomenínges se observan finas y descoloreadas por el incremento de la vascularidad. Microscópicamente los vasos del angioma son venas de pared fina, que varían en tamaño y se ocluyen por hialinización y proliferación endotelial, que se extienden por toda la lesión visible y a veces por la corteza subyacente y el plexo coroide ipsilateral.(4) Las calcificaciones se encuentran en las capas externas de la corteza cerebral y ocasionalmente se extienden hacia la sustancia blanca. Los depósitos de calcio se inician de manera adyacente a los vasos y de allí penetran al parénquima cerebral.(10) Es típica la atrofia adyacente al angioma, aunque no siempre es visible macroscópicamente. Microscópicamente se observan pérdida neuronal y gliosis que se extiende usualmente por toda el área afectada por el angioma.
Basado en los estudios del flujo sanguíneo cerebral y metabolismo energético, se ha postulado como hipótesis patogénica para la atrofia cerebral, el estado crónico de reducción de la perfusión sanguínea. Esta isquemia crónica puede explicarse por la obstrucción venosa producida en el angioma, que origina disminución del recambio sanguíneo en la zona. Los episodios agudos de hemiparesia asociados con crisis epilépticas están probablemente relacionados con trombosis venosas en áreas adyacentes al angioma. Resulta excepcional la presencia de hemorragias intracraneales en estos pacientes, lo que puede deberse a la escasa participación arterial en la formación angiomatosa.(4)
En la mayor parte de los casos, el control de las crisis epilépticas constituye el único elemento que se debe tratar; sin embargo, existen otras posibilidades y opciones que describiremos a continuación. En cuanto al tratamiento de la epilepsia, es importante la correcta selección de la droga antiepiléptica de acuerdo con el tipo o los tipos de crisis que muestra el paciente, y es necesario ser enérgicos por la importancia de las crisis en el pronóstico.(4) En escasos pacientes de corta edad, con afectación unilateral exclusivamente, con epilepsia refractaria y sin retraso mental profundo, está indicado el tratamiento quirúrgico, mediante la resección del área afectada, lobectomía e incluso hemisferectomía.(4,8) En estos casos, es importante asegurar que el otro hemisferio se encuentra totalmente sano, por lo que requiere una exquisita evaluación neuroimagenológica (estructural y funcional) y electroencefalográfica. Se ha propuesto el tratamiento con dosis bajas de aspirina para la prevención de la trombosis venosa y así del deterioro neurológico, sin embargo, esto es discutido.(3) En pacientes con lesiones faciales pequeñas y con escasa afectación neurológica, se puede utilizar laserterapia para el tratamiento estético.(3)
Con el registro de los casos descritos es posible que en un futuro próximo se pueda realizar una predicción de la gravedad y fenotipo para cada caso, así como el diseño de una terapéutica personalizada según criterios genéticos.
REFERENCIAS BIBLIOGRÁFICAS
1- Fernández Jaén A, Sánchez Jacobb R, Ramos Boludac E, et al. Síndrome de Sturge Weber con crisis epilépticas y calcificaciones intracraneales bilaterales en el periodo neonatal. Archivos de neurociencias [Internet]. 2004 [citado 14 May 2015];9:233-6. Disponible en: http://www.scielo.org.mx/scielo.php?script=sci_arttext&pid=S0187-47052004001200009&nrm=iso
2- Guevara Silva E, Garay Egashira M. Síndrome de Sturge Weber: Reporte de un caso y revisión de la literatura. Rev Diagnóstico [Internet]. 2011 [citado 14 May 2015];50(1). Disponible en: http://www.fihu-diagnostico.org.pe/revista/numeros/2011/ene-mar/44-47.html
3- Rios M, Barbot C, Pinto P, Salício L, Santos M, Carrilho I. Sturge-Weber syndrome-clinical and neuroimaging variability. An Pediatr [Internet]. 2012 [citado 14 May 2015];77(6):397-402. Disponible en: http://www.clinicalkey.es/#!/content/medline/2-s2.0-22743619
4- Suárez-Amor O, Cabanillas M, Monteagudo B, Ramírez-Santos A, de las Heras C. Síndrome de Sturge-Weber. An Pediatr [Internet]. 2010 [citado 14 May 2015];73(5):299. Disponible en: http://www.clinicalkey.es/#!/content/journal/1-s2.0-S1695403310003188
5- Montes AM, Toro AM, Arredondo MI, Arroyave JE, Vásquez LA, Molina V. Síndrome de Sturge-Weber tratado con láser de colorante pulsado. Rev Asoc. Colomb. Dermatol. Cir. Dermatol [Internet]. 2010 [citado 14 May 2015];18(4):236-8. Disponible en: http://search.ebscohost.com/login.aspx?direct=true&db=lth&AN=57523035&lang=es&site=ehost-live
6- Della Giovanna PS, Mauri ML, Boente MdC, Martin Loriente D, Fiandrino MJ. Síndrome de Sturge-Weber: estudio prospectivo. Dermatol Argen [Internet]. 2012;18(5). Disponible en: http://www.dermatolarg.org.ar/index.php/dermatolarg/article/view/887/535
7- García Jiménez J, Zubimendi Pérez S, Navarrete Páez M, Gutiérrez-Rojas L. Síntomas psicóticos en la enfermedad de Sturge-Weber. Psiq Biol [Internet]. 2013 [citado 14 May 2015];20(3):44-6. Disponible en: http://www.sciencedirect.com/science/article/pii/S1134593413000377
8- Parisi L, Di Filippo T, La Grutta S, et al. Sturge-Weber Syndrome: A Report of 14 Cases. Ment Illn. 2013;5(1). Citado en PubMed; PMID: 25478131.
9- Pila Pérez R, Conde Rivera ME, Pila Pélaez R, et al. Síndrome de Sturge-Weber: presentación de un caso. AMC [Internet]. 2010 [citado 14 May 2015];14(5). Disponible en: http://www.scielo.sld.cu/pdf/amc/v14n5/amc120510.pdf
10- Figueiredo LRC, Silva Filho FJd, Rehder JRCL. Síndrome de Sturge-Weber e suas repercussões oculares: revisão da literatura. Rev Bras de Oftal [Internet]. 2011 [citado 14 May 2015];70:194-9. Disponible en: http://www.scielo.br/pdf/rbof/v70n3/a14v70n3.pdf
11- Hospital Dr. Mario Muñoz Monroy de Colón. Historia Clínica No. 061223. Paciente LSH. Colón: Hospital Dr. Mario Muñoz Monroy de Colón; 2014.
Recibido: 17 de julio de 2015.
Aceptado: 24 de noviembre de 2015.
Meylín de la Caridad Morales Querol. Hospital Territorial Docente Dr. Mario Muñoz Monroy. Martí final. Colón. Matanzas, Cuba. Correo electrónico: meymor@fcmc.sld.cu

