










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Médica Electrónica
versión On-line ISSN 1684-1824
Rev.Med.Electrón. vol.40 no.3 Matanzas may.-jun. 2018
PRESENTACIÓN DE CASOS
Síndrome Rusell Silver. Presentación de caso
Russell-Silver syndrome. Presentation of a case
MSc. Elayne Esther Santana Hernández
Universidad de Ciencias Médicas de Holguín. Holguín, Cuba.
RESUMEN
El síndrome Russell Silver es una enfermedad genética de baja frecuencia, caracterizada por retardo del crecimiento prenatal y postnatal, dismorfias faciales y digitales, así como asimetría corporal. Se presenta una paciente femenina de dos años de edad, remitida a consulta de Genética Clínica, por retardo en el desarrollo pondoestatural psicomotor, dismorfias faciales y asimetría corporal. Se realizño el diagnóstico clínico de esta afección. Es importante establecer un diagnóstico precoz para la estimulación temprana, seguimiento multidisciplinario y se brindó un adecuado asesoramiento genético a los familiares.
Palabras clave: síndrome de Russel-Silver, hemihipertrofia, asimetría corporal, retraso del crecimiento intrauterino, hipocrecimiento con facie triangular y asimetria.
ABSTRACT
The Russell-Silver syndrome is a low-frequency genetic disease, characterized by a pre-natal growth retardation and postnatal digital and facial dysmorphia, and also body asymmetry. We present a female patient, aged 2 years, who was remitted to the consultation of Clinical Genetics because of a retardation in the psychomotor, height-weight development, facial dysmorphia and body asymmetry. The disease was clinically diagnosed. It is important to arrive to a precocious diagnosis for the early stimulation, multidisciplinary follow-up and adequate genetic advice to the relatives.
Key words: Russel–Silver syndrome, hemihypertrophy, body asymmetry, intrauterine growth retardation, stunted growth with triangular facies and asymmetry.
INTRODUCCIÓN
Este síndrome descrito por Silver en 1953 y Russell en 1964 como una entidad con manifestaciones clínicas variables, se describió gran expresividad variable en todos los casos reportados, lo que dificulta en ocasiones su identificación. Esta enfermedad se caracteriza por el hipocrecimiento pre y postnatal con facie triangular y asimetría corporal.1,2
Se plantean criterios diagnósticos en este síndrome, que se dividen en criterios mayores: retraso de crecimiento intrauterino, retraso de crecimiento postnatal, perímetro craneal normal, asimetría corporal y criterios menores: facie triangular, comisuras bucales dirigidas hacia abajo, mandíbula hipoplásica, malformaciones dentarias, clinodactilia, braquidactilia, sindactilia.3,4
El síndrome Russell Silver (SRS) se relaciona con múltiples alteraciones cromosómicas, tales como 47, XXY, en mosaicos X0/XY, incluso en trisomías 18 en mosaico y en deleción 8q. Se estima que esta afección se presenta cuando se producen otras muchas mutaciones genéticas y también por factores epigenéticos como la disomia uniparental materna del cromosoma 7.5
De esta enfermedad no se conoce su incidencia de forma precisa, solo se han documentado alrededor de 500 casos. Se estima una frecuencia mundial de un afectado por cada 100 000 nacidos vivos. En Cuba no se conoce la incidencia ni la frecuencia de esta enfermedad ya que existen muy pocos estudios publicados.
La mayoría de estos enfermos aparecen de forma esporádica en una familia, pero se han descrito genealogías, con varias personas afectadas con mayor o menor grado de manifestación del síndrome, las que siguen un patrón de herencia autosómico dominante con penetrancia incompleta; en menos ocasiones se ha sugerido una herencia autosómica recesiva, dominante ligada a X en otras, e incluso en una familia se ha visto una transmisión recesiva ligada a X.6
El objetivo de este trabajo es destacar lo importante que resulta delinear bien el fenotipo, para realizar un diagnóstico clínico precoz y brindar un adecuado asesoramiento genético a las familias. Lo que permitiría comenzar con la estimulación temprana y seguimiento multidisciplinario y elevara la calidad de vida de los afectados.
PRESENTACIÓN DEL CASO
Se presenta el caso clínico de una paciente femenina de APG remitida de su área de salud, a consulta de Genética Clínica del Centro Provincial de Genética de Holguín, por presentar retardo en el desarrollo pondoestatural psicomotor, dismorfias faciales y asimetría corporal.
Antecedentes prenatales: crecimiento intrauterino retardado.
Antecedentes perinatales: parto eutócico a las 39 semanas, peso 2 450 gramos, talla 48 cm, Apgar 8-9.
Antecedentes postnatales: de fallo de medro en el primer año al igual, y retardo en el desarrollo psicomotor. Por esta razón se atendió en consulta de neurodesarrollo, se le indico rehabilitación sosteniendo la cabeza a los 7 meses, se sentó al año, gateo y caminó a los 2 años.
Transicional: en el momento del estudio tenía 28 meses y se le realizó antropometría:
- Peso 9 200 g, peso/edad por debajo del tercer percentil.
- Talla 76 cm, talla /edad entre el tercer y el décimo percentil.
- Circunferencia cefálica 46 cm en el 97 percentil. Peso/ talla en el 10 percentil.
Examen físico
Cabeza: cráneo con ligera braquicelia y fontanela anterior amplia de aproximadamente 2 cm.
Cara: triangular con mentón estrecho y pequeño, con retromicrognatia, frente ancha y amplia con implantación baja del cabello, con cejas muy pobladas, ligero hirsutismo. Asimetría facial con la hemicara derecha más pequeña, llegando a tener más descendida la región orbital de ese lado con asimetría de las hendiduras palpebrales. Comisura labial hacia abajo, orejas grandes despagadas, en rotación posterior. Estas características se pueden apreciar en la figura 1.

Extremidades superiores: cortas con el primer dedo grueso y de implantación alta bilateral, así como clinodactílea del quinto dedo.
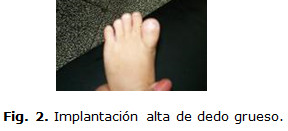
Extremidades inferiores: con el primer dedo de ambos pies gruesos de implantación alta, acompañado de clinodactilea del quinto dedo. Lo que se puede observar en la figura 2.
Tórax: configuración normal. Soplo sistólico grado I/VI.
Se le realizó ecocardiograma y no se detecto ninguna alteración estructural. Se determinó que presentaba soplo funcional.
Abdomen: globuloso depresible que sigue los movimientos respiratorios. No visceromegalia.
Hemicuerpo izquierdo más grande que el lado derecho. Donde la extremidad superior e inferior se midió y el lado izquierdo es 2 cm más largo.
Genitales externos: de configuración normales.
Se realizaron los siguientes estudios:
- Ecografía abdominal: normal.
- Cariotipo que resultó en 18 metafases: 46, XX una hembra cromosómicamente normal.
- Perfil tiroideo: normal.
- Hemograma completo: normal.
- Estudios bioquímicos con perfil hepático, proteínas y lípidos: normales.
- Radiografía de los miembros superiores e inferiores: se comprobó por la medida de los huesos largo de húmero y fémur izquierdo más largos que los derechos.
- Radiografía de ambas manos determinando edad ósea retrasada.
DISCUSIÓN
El síndrome de Silver Russel (SRS) es una entidad con unas manifestaciones clínicas y alteraciones genéticas muy heterogéneas donde no suele faltar un importante retraso en el crecimiento pre y postnatal, asimetría corporal con un fenotipo facial típico (cara triangular) y relativa macrocefalia.1
En el SRS la mayoría de los casos son esporádicos, sin embargo se ha descrito la aparición de casos familiares lo que ha sugerido la posibilidad de transmisión hereditaria. Al igual que la variedad de manifestaciones en la presentación clínica, en el SRS existe un amplio espectro de mutaciones y alteraciones en las bases genéticas que justifican su aparición.2,3
Se ha relacionado con diferentes alteraciones cromosómicas: cariotipo 47XXX; translocaciones 17;20 y 1;17; deleciones: 8q11-q13; y 17q22-q24, deleción del brazo corto del cromosoma 18 y mosaicos trisomía 18/normal o triploidía/normal, mixoploidías.4
También la pérdida de metilación paterna en el H19-IGF2 (ICR1) puede ser la causa subyacente en un número importante de casos de SRS. La disomía uniparental materna del cromosoma 7 (UPD7) es la causa en el 10 % de los casos.6,7
Se describen factores epigenéticas como el que se refiere al 35 % de pacientes con SRS que afectan a la impronta de la región cromosómica 11p15.5, región que guarda una estrecha relación con el síndrome de Beckwith Wiedemann.7
En esta región se han descrito mutaciones puntuales en distintos genes implicados, como son: GRB10 (7p11-p12), MEST (7q32), IGF2 ICR1, y H19 (11p15).8
El diagnóstico diferencial es muy importante teniendo en cuenta la asimetría corporal, considerándose como importantes para descartar las siguientes entidades clínicas: tumores primarios renales (tumor de Wilms), hepáticos, tumor de células adrenales, síndrome de Beckwith-Wiedmann, Klippel-Trenaunay-Weber y McCune-Albright; todos ellos con características especiales que requieren estudios adicionales.7-10
Lo ocurrido en esta paciente se consideró como caso esporádico, por nueva mutación, como la mayoría de los casos reportados en la literatura. El reporte de este caso resalta la importancia del diagnóstico precoz, para establecer estimulación temprana, con adecuado seguimiento multidisciplinario en conjunto, donde confluyan especialistas de Pediatría, Nutrición, Fisioterapia y Rehabilitación, Ortopedia y Genética Clínica para mejorar la calidad de vida de los afectados, además de brindar un adecuado asesoramiento genético a los familiares.
REFERENCIAS BIBLIOGRÁFICAS
1- Õunap K. Silver-Russell Syndrome and Beckwith-Wiedemann Syndrome: Opposite Phenotypes with Heterogeneous Molecular Etiology. Mol Syndromol. 2016 Jul;7(3):110-21. Citado en PubMed; PMID: 27587987.
2- Giabicani E, Netchine I, Brioude F. New clinical and molecular insights into Silver-Russell syndrome. Curr Opin Pediatr. 2016 Aug;28(4):529-35. Citado en PubMed; PMID: 27386972.
3- Goto M, Kagami M, Nishimura G, et al. A patient with Temple syndrome satisfying the clinical diagnostic criteria of Silver-Russell syndrome. Am J Med Genet A. 2016 Sep;170(9):2483-5. Citado en PubMed; PMID: 27362607.
4- Wakeling EL, Brioude F, Lokulo-Sodipe O, et al. Diagnosis and management of Silver-Russell syndrome: first international consensus statement. Nat Rev Endocrinol. 2017 Feb;13(2):105-124. Citado en PubMed; PMID: 27585961.
5- Luk HM, Yeung KS, Wong WL, et al . Silver-Russell syndrome in Hong Kong. Hong Kong Med J. 2016 Dec;22(6):526-33. Citado en PubMed; PMID: 27468965.
6- Ishida M. New developments in Silver-Russell syndrome and implications for clinical practice. Epigenomics. 2016 Apr;8(4):563-80. Citado en PubMed; PMID: 27066913.
7- Jurkiewicz D, Kugaudo M, Skórka A, et al. A novel IGF2/H19 domain triplication in the 11p15.5 imprinting region causing either Beckwith-Wiedemann or Silver-Russell syndrome in a single family. Am J Med Genet A. 2017 Jan;173(1):72-78. Citado en PubMed; PMID: 27612309.
8- Carrera IA, De Zaldívar MS, Martín R, et al. Microdeletions of the 7q32.2 imprinted region are associated with Silver-Russell syndrome features. Am J Med Genet A. 2016 Mar;170(3):743-9. Citado en PubMed; PMID: 26663145.
9- Ríos-Méndez RE, Montero-Monar HE, Pernández-Alvarado AP, et al. Silver-Russell syndrome (hemihypertrophy) and cor triatriatum in a newborn. Arch Argent Pediatr. 2015 Jun;113(3):e140-4. Citado en PubMed; PMID: 25996332.
10- Azzi S, Salem J, Thibaud N, et al. A prospective study validating a clinical scoring system and demonstrating phenotypical-genotypical correlations in Silver-Russell syndrome. J Med Genet. 2015 Jul;52(7):446-53. Citado en PubMed; PMID: 25951829.
Recibido: 25/10/16
Aprobado: 5/4/18
Elayne Esther Santana Hernández. Universidad de Ciencias Médicas de Holguín. Avenida Lenin No. 4 esquina Aguilera. Holguín, Cuba Correo electrónico: esantana@hpuh.hlg.sld.cu

