










 Servicios personalizados
Servicios personalizados
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
INTRODUCCIÓN
La incontinencia pigmentaria (IP) o síndrome de Bloch-Sulzberger, es una genodermatosis de escasa frecuencia, ligada en forma dominante al cromosoma X, consecuencia de una mutación en el gen IKBKG/NEMO, localizado en el Xq28. Codifica un inhibidor del potenciador del gen del polipéptido ligero kappa en células B Cinasa Gamma, el cual es un modulador esencial del factor de transcripción nuclear kappa B. Tiene una prevalencia de 1/50000 recién nacidos vivos y una incidencia de 0,7 en 100 000 nacimientos, con 27,6 casos nuevos por año en el mundo, aunque se estima pueda ser mayor, debido a que las lesiones dermatológicas pueden pasar desapercibidas o confundirse con otras entidades.1-5
Las manifestaciones cutáneas están siempre presentes y se dividen en cuatro fases.1-5 Afecta la pigmentación de la piel y suele estar asociada con una gran variabilidad de alteraciones en los ojos, las uñas, el pelo, los dientes, el tejido óseo, el sistema cardiovascular y el sistema nervioso central.1-13
Se ha descrito y observado una alta penetrancia y expresión clínica muy variable en miembros de una misma familia. Algunas pacientes solo presentan síntomas y signos cutáneos, mientras que otras pueden sufrir hasta una severa incapacidad neurológica y/o oftalmológica.1-6,14-16
Teniendo en cuenta la dificultad diagnóstica que en ocasiones ofrece esta enfermedad en sus etapas iniciales, dada su similitud con otras dermatosis, se presenta una lactante de 1 mes, por mostrar desde su nacimiento manifestaciones en piel de eritema y vesicoampollas de contenido seroso y turbio diseminadas. El aspecto de las lesiones, su distribución y su evolución, además de los antecedentes de afecciones cutáneas similares en la infancia -en dos miembros de la familia materna-, permitió establecer el diagnóstico de IP, con colaboración del Servicio de Genética, Neuro-Oftalmología y Anatomía Patológica. Se resaltan las tres primeras fases cutáneas de esta enfermedad en la niña, las manifestaciones oftálmicas y la eosinofilia periférica, por lo que constituye un caso de IP familiar.
PRESENTACIÓN DEL CASO
Motivo de consulta: lesiones en la piel.
Historia de la enfermedad actual: lactante ELlD, blanca, femenina, de 1 mes de edad, de procedencia rural, que inicia con lesiones en piel desde la primera semana de vida, a predominio acral, valorada en su área de salud, donde se diagnostica con eritema tóxico del recién nacido. Al continuar apareciendo lesiones, es evaluada nuevamente en su consultorio, y se impone tratamiento para escabiosis a ella y al resto de los convivientes, con permetrina 5 % (crema). Según refiere la madre, las lesiones empeoraron y aparecen nuevas en la región superior del tronco, sin presentarse en otros miembros de la familia. Por tal motivo es traída a consulta hospitalaria, donde se decide ingreso para estudio y tratamiento.
APP: parto eutócico a 38,3 semanas; apgar 9/9; peso 3 040 g; fibroma preauricular izquierdo.
APF: madre: lesiones cutáneas desde su nacimiento sin diagnóstico, que perduraron hasta la pubertad en forma de “manchas oscuras”. Agenesia renal. Aborto espontáneo en primer trimestre sin determinar sexo. G3P2A1 (espontáneo). Prima materna con lesiones de piel similares en la infancia.
Examen físico al ingreso: vesicoampollas pequeñas, de contenido turbio, sobre base eritematosa, que siguen un patrón lineal, que alternan con pápulas, de distribución diseminadas en tronco y miembros superiores e inferiores, siguiendo las líneas de Blaschko. (Fig. 1 y fig. 2)
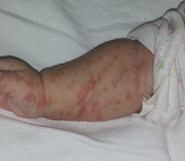

Al ingreso por presentar lesiones con signos de piodermitis, se impone tratamiento con baños antisépticos (permanganato de potasio), antibiótico sistémico (cefazolina IM) y crema antibiótica en lesiones costrosas (gentamicina). Se indican estudios complementarios y se solicita interconsulta con Genética, Neuro-Oftalmología y Cardiología.
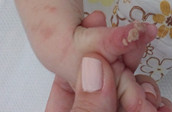
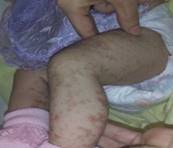
Durante los 15 días de estadía hospitalaria, se observa cómo las lesiones transitan por las tres primeras fases de la enfermedad: la inflamatoria (descrita al examen físico del ingreso); la hiperqueratósica, con lesiones de aspecto verrugoso, de base eritematosa, con bordes definidos, superficie rugosa, de 0,5-1 cm de diámetro, que conservan su individualidad, escasas en número, de distribución localizada en primer dedo de mano derecha y primer artejo de pie izquierdo (fig. 3 y fig. 4), y, por último, la fase hiperpigmentada, caracterizada por lesiones hipercrómicas de bordes no bien definidos, arremolinadas y siguiendo patrón lineal, de distribución diseminada a predominio de tronco y extremidades. (Fig. 5)

Resultados de complementarios: hemoglobina: 13,0 g/L; eritrosedimentación: 12 mm/s; leucograma: 11,2 x 109 (eosinófilos: 10,8). El resto de la analítica se encontraba dentro de los parámetros normales.
Ecocardiograma: sin alteraciones estructurales.
Neurooftalmología: distrofia de la retina (albinismo ocular +) e hipoplasia bilateral del disco óptico. Se indica tomografía axial computarizada para descartar alteraciones orgánicas, pero los padres se niegan a dicho estudio.
Genética: se confecciona el árbol genealógico; se brinda consejo genético a la familia y se mantiene seguimiento de la paciente. Por tratarse de una lactante de 1 mes de vida no se puede determinar aún alteraciones en el desarrollo psicomotor, esfera cognitiva y trastornos dentarios.
Ultrasonido abdominal y Rx de tórax: no existen alteraciones orgánicas.
Biopsia de piel: infiltrado inflamatorio con abundantes eosinófilos, vesículas intraepidérmicas, espongiosis, infiltrado inflamatorio crónico de la dermis superior, melanófagos cargados de melanina, correspondiente a incontinencia pigmentaria.
Actualmente, se mantiene en seguimiento en consulta multidisciplinaria, manteniendo lesiones pigmentadas arremolinadas, que siguen las líneas de Blaschko de distribución diseminada.
DISCUSIÓN
La IP es un trastorno neuroectodérmico sistémico, caracterizado por lesiones cutáneas que evolucionan por estadios: vesiculoso-pustuloso, verrugoso, hiperpigmentado e hipopigmentado. Estas lesiones en la piel desaparecen y evolucionan espontáneamente en el tiempo.1-9 Se asocian con compromisos oftalmológicos (30 %), neurológicos (5 a 13 %) y odontológicos (30 a 40 %), que son permanentes.6-16 Es una entidad muy rara, con una prevalencia de 1/50 000 nacidos vivos, y se manifiesta casi exclusivamente en mujeres, ya que la mayor parte de los varones mueren in útero y sobreviven si su cariotipo es 47XXY, o si presentan una mutación hipomórfica. Tiene un patrón de herencia autosómico dominante, ligado al cromosoma X (Xq28).1-18
La IP, antes conocida como síndrome de Bloch-Sulzberger, es una genodermatosis poco frecuente. Fue descrita por Garrod en 1906, y posteriormente definida por Bloch (1926) y Sulzberger (1928) como Incontinentia pigment, citado por Landy1) Enei2) y Romero et al.,3) basados en los hallazgos histopatológicos característicos (aunque no patognomónicos) de las lesiones cutáneas de la tercera fase de la enfermedad, en la cual se encuentra melanina libre en la dermis, o dentro de los melanófagos dérmicos.4-18
En su etiopatogenia se invoca que el factor de transcripción nuclear kappa B, es activado por la proteína producto del gen IKBKG/NEMO, esencial modulador que desempeña un importante papel en múltiples funciones fisiológicas, como las respuestas inmunes y el estrés, las reacciones inflamatorias, el desarrollo ectodérmico, la adhesión tisular y la protección celular contra la apoptosis inducida por el factor de necrosis tumoral. La mutación de este gen conduce a la disminución del factor de transcripción nuclear kappa B, provocando un aumento de la susceptibilidad de las células a la apoptosis. Por ello en el sexo masculino la apoptosis extensa es responsable de la muerte fetal temprana, así como de alteraciones a nivel hepático. Las reacciones inflamatorias y el reclutamiento epidérmico de eosinófilos, observado en la primera etapa, puede estar relacionado con una quimiocina selectiva de eosinófilos (eotaxina), producida por leucocitos específicos, como eosinófilos, macrófagos y células T, y por algunas células estructurales, como las endoteliales, los fibroblastos y las epiteliales.3-9
Las manifestaciones clínicas de la IP son variadas, incluso en miembros de la misma familia.4 Existe una estrecha relación de la severidad de la entidad por la asociación de manifestaciones oftalmológicas, neurológicas y recurrencias de brotes en las lesiones en la piel.1) En la paciente se describen estas asociaciones de comienzo temprano (antes de los 2 meses), con signos oftalmológicos demostrados, por lo que se cumplen los criterios mayores de IP propuestos por Landy y Donnai1) en 1993, y su actualización por Mink , Chen y otros en 2014 y 2015.14-20
Los diagnósticos diferenciales que se deben descartar ante una sospecha de IP van a depender de la etapa en la que se encuentre la enfermedad: para la fase vesicoampollar cabe plantear herpes neonatal, impétigo ampolloso por Staphylococcus aureus, mastocitosis, histiocitosis, epidermólisis bulosa hereditaria y enfermedad ampollar por IgA lineal. Para el estadio verrugoso o hiperqueratósico, se deben incluir nevus epidérmico lineal y liquen estriado. Cuando existe predominio de hiperpigmentación, cabe señalar la hipermelanosis nevoide lineal y en remolinos, así como el síndrome de Naegeli-Franceschetti-Jadassohn.7-18
En el caso que se presenta, es válido resaltar la evolución acelerada de la enfermedad con la existencia de las tres primeras fases antes de los dos meses de vida, con afectación oftálmica demostrada, presentando mayor severidad en esta generación, pues la madre y prima materna solo manifestaron signos cutáneos hasta la adolescencia, sin repercusión en otros sistemas.
En la mayor parte de los pacientes, el inicio temprano de múltiples expresiones clínicas (antes del primer año) ensombrece el pronóstico, por lo que la detección precoz es importante, y aunque no existe en la actualidad un tratamiento curativo, la conducta ante estos casos es sintomática e interdisciplinaria, atendiendo al sistema afectado.1-11 En el caso expuesto, el seguimiento se mantiene por Dermatología, Neuro-oftalmología, Genética y Pediatría. Se indica valoración y seguimiento por Fisiatría y Estomatología para pesquisaje de posibles alteraciones en estos sistemas.
La IP es una genodermatosis rara y poco frecuente, con expresión fenotípica variable en una misma familia, cuya severidad depende de la proporción de los brotes de las lesiones en la piel y del inicio temprano de alteraciones orgánicas. La afectación es multisistémica y su pronóstico reservado. El diagnóstico debe ser precoz y su tratamiento multidisciplinario. El asesoramiento genético familiar resulta crucial ante la presencia de un patrón de herencia ligado al X.5-6,14-20

