










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Informática Médica
versión On-line ISSN 1684-1859
RCIM vol.5 no.1 Ciudad de la Habana ene.-jun. 2013
ARTÍCULO ORIGINAL
Herramienta web para la clasificación de microsatélites polimórficos en genomas bacterianos
Web Tool for classfication of polymorphic microsatellites in bacteria genomes
MsC. Carlos M. Martínez Ortiz,I MsC. Miguel Sautié Castellanos,II Dra. Yordanka Cuza Ferrer,III Ing. Yinette Wisdom ViñaIV
IProfesor Auxiliar. Lic. Microbiología y Máster en ciencias en Bioquímica de las proteínas. Centro de Cibernética Aplicada a la Medicina (CECAM). La Habana, Cuba. E-mail: cmmo@infomed.sld.cu
IIProfesor Auxiliar. Lic. Bioquímica. Máster en ciencias en Informática Médica. Centro de Cibernética Aplicada a la Medicina (CECAM). La Habana, Cuba. E-mail: msc@infomed.sld.cu
IIIProfesora Asistente. Dra. en Medicina. Especialista en Fisiología Normal y Patológica. ICBP "Victoria de Girón". La Habana, Cuba. E-mail: yordankacuza@infomed.sld.cu
IVProfesora Instructor. Ingeniera en Informática. Producciones Trimagen. S.A. La Habana, Cuba. E-mail: yinette@trimagen.co.cu
Las secuencias repetidas en tándem, específicamente los mini y micro satélites, han demostrado ser muy eficaces en la clasificación de bacterias patogénicas como B. anthracis, M. tuberculosis y P. aeruginosa, entre otras. En humanos es manifiesta su participación estando relacionados con más de ochenta enfermedades, gran parte de ellas de tipo neurodegenerativas, musculares y algunos tipos de cáncer. La herramienta web que presentamos es el resultado de la detección computacional de estas secuencias en genomas bacterianos completos y su correspondiente anotación en la estructura genómica de acuerdo a las diferentes regiones donde estos se localizan. La herramienta tiene como fin primario brindar un sistema relacional que permita al investigador ubicar los microsatélites de diferentes especies bacterianas, con más de un genoma secuenciado para inferir su posible carácter polimórfico, dentro del contexto de la estructura genómica y así proveer un primer acercamiento al rol putativo que los microsatélites desempeñan desde el punto de vista funcional. La herramienta se puede aplicar no solo en estudios taxonómicos y epidemiológicos sino en la detección de posibles relaciones de estas secuencias con las funciones moleculares, procesos biológicos y, en última instancia, las diversas formas de evolución de estas especies. El sitio web brinda el servicio de consultas a la base de datos de microsatélites bacterianos de acuerdo al sistema de tablas relacionales y atributos propios de las mismas. Cuenta además con los servicios típicos de un sitio con estas características como: sistema de autenticación, foro, encuestas, enlaces y documentación sobre la metodología empleada y del tema en cuestión.
Palabras clave: microsatélites, repetidos en tándem, bacterias, sistema de base de datos.
ABSTRACT
The tandem repeat sequences, especially mini and microsatellites, have proven to be very effective in classification of pathogenic bacteria such as B. anthracis, M. tuberculosis and P. aeruginosa, among others. In human beings it is manifest its participation, being related with over eighty diseases, nearly all neurodegenerative and muscular, and some kinds of cancer. The web tool we are offering here is the result of computational detection of these sequences in whole bacteria genomes, and its respective annotation in the genomic structure according to the different regions where they are localized. The primary goal of this tool is to offer a relational system that allows mapping the microsatellites of bacterial species, all of them with more than one genome sequenced to infer their possible polymorphic character, in the context of genomic structure and thus providing a first approach to the putative role they perform from the functional point of view. The tool can be applied not only in taxonomical and epidemiological studies but in the detection of possible relationships of these sequences with the molecular functions, the biological processes and, as a last resort, the different forms of these species evolution. The web site offers the service of queries to the bacterial microsatellites database according to the related tables and its inherent attributes. It also has the typical services of this kind of site like: logging system, forum, polls, links and documentation about the employed methodology and the topic.
Key words: microsatellites, tandem Repeat, bacteria, data base system.
INTRODUCCIÓN
El descubrimiento del ADN satélite en 19611 inició el estudio de las secuencias repetidas en tándem develándose con el tiempo un amplio espectro de estas secuencias en cuanto a composición, tamaño y localización genómica.
Estos marcadores genéticos han demostrado ser muy útiles constituyendo el elemento clave en las pruebas forenses para la identificación de personas y animales. Los Repetidos en Tándem de Longitud Variable (VNTR por sus siglas en inglés) son de vital importancia en los estudios genéticos de linaje pues, a diferencia de los polimorfismos de simple nucleótido (SNP), exhiben más de un alelo con alta frecuencia en el número de copias, conduciendo así a altas tasas de heterocigosis.2
Desde hace más de 18 años se conoce la participación de los repetidos en tándem (RTs) como agentes causales de enfermedades en humanos. Dentro de las más conocidas se encuentran la atrofia muscular espinobulbar, la enfermedad de Huntington y las ataxias espinocerebelosas de tipo 1, 2, 3, 6 y 7, todas relacionadas con la expansión del triplete CAG en regiones codificantes. Asociadas a expansiones en regiones no codificantes están el síndrome de Fragil X, la ataxia de Friedreich, la distrofia miotónica y las ataxias espinocerebelosas de tipo 8 y 12.
Los repetidos en tándem, particularmente los mini y microsatélites, se encuentran también en organismos procariotas y han sido de gran utilidad en estudios de epidemiología molecular.3 En bacterias patógenas, los RTs fueron inicialmente identificados asociándose a genes causantes de la virulencia. Las técnicas que emplean los RTs como marcadores han sido efectivas donde otras, de carácter molecular inclusive, han fallado.4-5 Por ejemplo, la técnica de clasificación de secuencias multilocus (MLST)6 actual referencia en epidemiología molecular para Nisseria meningitidis, no es aplicable en varios tipos de gérmenes como son B. anthracis, M. tuberculosis y Y. pestis, debido al reciente surgimiento de estos patógenos y su consecuente variabilidad en las secuencias. En estos casos los RTs han resultado marcadores muy informativos para la clasificación genética de estas especies. La contribución de los RTs al polimorfismo genómico ha quedado establecida por diseños como el AFPL (polimorfismo en fragmentos de longitud amplificada), quedando ilustrada claramente en B. anthracis, donde se demostró que las bandas polimórficas en los patrones de AFPL se debían a variaciones de secuencias repetidas en tándem.
Los RTs exhiben una alta tasa de mutación debido a una variedad de mecanismos que afectan su estabilidad entre los que se encuentran el deslizamiento en la replicación y el entrecruzamiento desigual en la meiosis.7
En internet existen publicados varios sitios que brindan servicios de consulta en repositorios de secuencias repetidas en tándem. Estos repositorios han sido construidos algunos para aplicaciones muy específicas, varios de ellos dedicados precisamente a organismos bacterianos,8-9 y otros de carácter general incluyendo todo tipo de especies.2
La creación de algoritmos para detectar secuencias repetidas en tándem ha sido un tema muy abordado en la literatura y continúa siendo un problema computacional si tenemos en cuenta el crecimiento exponencial que exhiben los bancos de secuencias. Estos algoritmos se pueden clasificar de acuerdo a tres esquemas generales. El primer grupo emplea un esquema puramente combinatorio que recorre la secuencia linealmente y selecciona los RTs de acuerdo con determinadas reglas de construcción de estas secuencias. El segundo grupo usa criterios probabilísticos para seleccionar RTs candidatos que luego son sometidos a pruebas de evaluación para su selección final. El tercer esquema utiliza el alineamiento con patrones o librerías de estos y los RTs seleccionados son aquellos que obtienen una puntuación por encima de determinado valor de corte.10-13
La herramienta web que presentamos hace uso de una base de datos relacional (MSB_DB) permitiendo consultar la información de los microsatélites (localización, tamaño, unidad repetida, etc) relacionada con gran parte de las anotaciones que presentan los ficheros de secuencias genómicas del GeneBank, (ej. organism, gene, CDS, RNA, etc.). Los genomas bacterianos escogidos fueron los de aquellas especies que poseían más de un genoma secuenciado lo que permite hacer inferencias sobre el carácter polimórfico de los microsatélites contenidos en ellos.
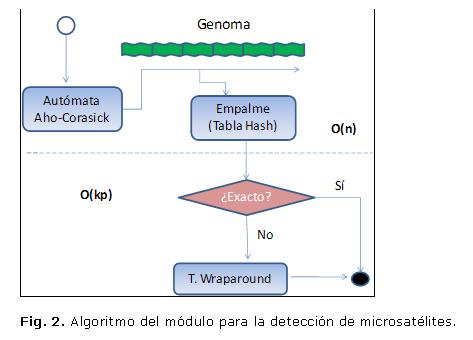
Se diseñó un algoritmo para la detección de los microsatélites que emplea un esquema combinado de detección exacta de todas las ocurrencias de patrones mediante el autómata Aho-Corasick y de extensión aproximada de los mismos mediante alineamiento wraparound, aplicando una distribución probabilística como criterio de parada. El algoritmo es eficiente y aplica además determinadas reglas heurísticas para seleccionar los microsatélites candidatos.
MATERIALES Y MÉTODOS
Las secuencias genómicas de bacterias con más de un genoma secuenciado fueron extraídas del sitio ftp://ftp.ncbi.nih.gov/genomes/Bacteria/-all.gbk.tar.gz. En este archivo se encuentran todas las secuencias genómicas de bacterias en formato de ficheros planos GenBank (GBFF). Los ficheros planos son fáciles de acceder, distribuir y además de mantener. La mayoría de las aplicaciones para análisis de secuencias tienen herramientas para su utilización. En nuestro caso empleamos el paquete Biojava para extraer la información en ellos contenida.
Para la creación de la base de datos MSB_DB, se implementó una aplicación en Java (JDK 6) la cual incluyó tres módulos fundamentales: I) módulo empleando el analizador sintáctico contenido en el paquete Biojava 1.7, II) módulo empleando el API JDBC para acceso a bases de datos, en este caso se empleó el gestor de bases de datos MySQL y III) módulo para la detección de microsatélites que fue implementado en lenguaje C++.
Para la creación del sitio web se empleó el gestor de contenidos DRUPAL 6.3 y para la gestión interna de la base de datos MSB_DB del sitio se programaron scripts para consultas de actualización y de selección en el lenguaje PHP 5.3. Como gestor de base de datos se empleó MySQL 5.5. El servicio http corrió a cargo de Apache 2.2.
RESULTADOS Y DISCUSIÓN
En la figura 1 se muestra el algoritmo general para la generación de la base de datos MSB_DB. Este transcurre a través de dos módulos principales que tienen como entrada los ficheros planos del GenBank. El primero fue programado en Java y tiene la función de extraer rasgos y anotaciones. El segundo fue programado en C++ y tiene la función de detectar y extraer los microsatélites presentes en las secuencias genómicas. Ambos módulos contribuyen con sus salidas al tercer módulo que se encarga de actualizar la base de datos MSB_DB y establecer las relaciones necesarias.

El módulo de detección de microsatélites (Fig. 2), realiza primeramente la búsqueda exacta y exhaustiva, a lo largo de toda la secuencia de tamaño n, de todas las ocurrencias de todos los patrones posibles de tamaño entre 1 y 8 nucleótidos. Esto lo hace mediante el algoritmo de Aho-Corasick, autómata que dado un diccionario de palabras detecta todas las ocurrencias de estas en un texto. Luego empalma estas ocurrencias cuando son del mismo patrón y están unas consecutivas a las otras y reporta la posición, longitud y composición del repetido. Posteriormente, si se elige la opción de hallar repetidos aproximados se realiza la extensión de las secuencias antes reportadas mediante alineamiento wraparound. Para el caso de los microsatélites registrados en esta base de datos se emplearon los siguientes parámetros para el alineamiento: match=2, mismatch=-4, indel=-4, flank=3. El parámetro flank es un factor que multiplicado por el tamaño del repetido exacto obtenemos las secuencias a considerar en los flancos para hacer el alineamiento local wraparound, el resto de los parámetros son los referidos a coincidencias, no coincidencias e inserción-deleción y son comunes a cualquier tipo de alineamiento. El algoritmo es muy eficiente: O(n) en la primera fase y O(kp) en la fase de extensión, donde k es la longitud del repetido candidato y p la longitud del patrón. El algoritmo emplea además determinadas reglas heurísticas que agilizan aún más la búsqueda y sesgan el número de microsatélites candidatos. El módulo tiene una versión standalone que puede ser descargada del sitio y en la que el usuario puede emplear parámetros de búsqueda personalizados.
En la figura 3 se muestra el esquema de la base de datos MSB_DB. La tabla TLocus registra anotaciones generales de cada genoma y cada entrada tiene múltiples entradas en la tabla TReference que registra las diferentes referencias bibliográficas relacionadas con la secuenciación de dichos genomas. La tabla TFeature registra una selección de los rasgos anotados y su localización en la secuencia genómica, cada entrada de esta tabla tiene múltiples entradas a la tabla TAnnotation que registra las anotaciones hechas a los rasgos en forma de pares clave-valor. La tabla TRepeat registra los microsatélites propiamente: su posición en la secuencia, la unidad repetida, el número de estas unidades y si es exacto o no. Una registro de esta tabla puede tener múltiples entradas en la tabla TAligment donde se caracteriza el alineamiento de este microsatélite con la unidad repetida. La relación entre TFeature y TRepeat es de muchos a muchos y esto es debido a que en el tramo de secuencia relativo a un rasgo pueden existir muchos microsatélites distintos pero a su vez un mismo microsatélite puede estar en diferentes rasgos anotados. Esto se debe a redundancia en la anotación de rasgos en la cual un rasgo específico puede estar embebido en un rasgo más general.

En la figura 4 se muestra una vista general del sitio y la página generada luego de hacer una consulta de selección que relaciona las tablas TLocus, TFeature y TRepeat. En esta consulta, truncada por la vista de browser, se pueden apreciar algunas entradas que dan fe precisamente del cumplimiento de uno de los objetivos propuestos con la creación de esta base de datos: el de detectar y clasificar microsatélites polimórficos. Se puede observar como las entradas para los genes hemX y kpsE presentan microsatélites polimórficos que varían en longitud siendo las mismas unidades repetidas y estando presente en diferentes genomas de la misma especie, en este caso Escherichia coli. Esta especie es una de las más estudiadas debido al desempeño que ha tenido en el desarrollo de la biología molecular y por tener varios serotipos reconocidos como agentes patógenos en humanos, por ejemplo: E. coli O157:H7, E. coli O121 y E. coli O104:H21. En nuestra base existen más de 30 secuencias genómicas de esta especie lo cual la hace una magnífica candidata para el estudio del polimorfismo presente en microsatélites bacterianos.
El sitio cuenta con un menú de primer nivel que da entrada a cinco páginas principales: Inicio, MSB_Select, MSB_Update, Foro y Encuesta. En la página Inicio encontramos documentación sobre el sitio y enlaces a otras documentaciones relacionadas con la metodología del trabajo. Cuenta además con un sistema de autenticación que permite clasificar a los usuarios de acuerdo a determinados privilegios. Por ejemplo, los usuarios con el rol de administrador pueden realizar consultas de modificación sobre la bases de datos, funcionalidad que está vedada para el resto de los usuarios. Los usuarios anónimos tienen una navegación limitada sin poder hacer consultas de selección ni participar en los temas de foros.
CONCLUSIONES
La herramienta presentada cumple con el propósito original que nos planteamos, que fue el de crear un recurso bioinformático que permitiera la clasificación de los microsatélites presentes en genomas bacterianos. La base de datos MSB_DB permite la clasificación de estas secuencias y además relacionarlas directamente con las anotaciones presentes en los bancos de secuencia primarios. Esto nos permite tener un primer acercamiento a la función de los microsatélites en el contexto de la estructura del genoma en sus niveles génico y subgénico. El esquema de la base de datos nos permite detectar y clasificar, dentro del contexto de rasgos y anotaciones, microsatélites polimórficos presentes en variantes de las mismas especies y en los mismos locus genéticos. Al estar basada en tecnología web garantiza mayores niveles de acceso a la misma por parte de la comunidad científica. El sitio fue creado en su totalidad con herramientas de software libre, es intuitivo, de fácil navegación y puede ser enriquecido a partir de las encuestas, comentarios y temas de foro. En este momento se encuentra público en la intranet de nuestra institución en espera de ser hospedado en una red de mayor acceso. Como trabajo futuro nos proponemos ampliar la base de datos creando nuevas relaciones con otros repositorios que brinden información más específica sobre los procesos biológicos y las funciones moleculares en que participan los genes anotados en los bancos primarios de genomas.
REFERENCIAS BIBLIOGRÁFICAS
1. Kit S. Equilibrium sedimentation in density gradients of DNA preparations from animal tissues. J. Mol. Biol. 1961; 3: 711-716.
2. Gelfand Y, Rodriguez A, Benson G. TRDB-The Tandem Repeats Database. Nucleic Acids Research. 2007; 35, Database issue doi:10.1093/nar/gkl1013.
3. van Belkum. High-throughput epidemiologic typing in clinical microbiology. Clin Microbiol Infect A. 2003; 9:86-100.
4. Radomski N, Thibault VC, Karoui C, de Cruz K, Cochard T, Gutierrez C, Supply P, Biet F, Boschiroli ML. Determination of genotypic diversity of Mycobacterium avium subspecies from human and animal origins by mycobacterial interspersed repetitive-unit-variable-number tandem-repeat and IS1311 restriction fragment length polymorphism typing methods. J Clin Microbiol. 2010 Abr;48(4):1026-34.
5. Guo C, Liao Y, Li Y, Duan J, Guo Y, Wu Y, Cui Y, Sun H, Zhang J, Chen B, Zou Q, Guo G. Genotyping analysis of Helicobacter pylori using multiple-locus variable-number tandem-repeats analysis in five regions of China and Japan. BMC Microbiol. 2011 Sep 3;11:197.
6. Vergnaud G, Pourcel C. Multiple locus variable number of tandem repeats analysis. Methods Mol Biol. 2009;551:141-58.
7. Bichara M, Wagner J, Lambert IB. Mechanisms of tandem repeat instability in bacteria. Mutation Research. 2006; 598: 144-163.
8. Le Fiache P, Hauck Y, Onteniente L, Prieur A, Denoeud F, Ramisse V, Sylvestre P, Benson G, Ramisse F, Vergnaud G. A tandem repeats database for bacterial genomes: application to the genotyping of Yersinia pestis and Bacillus anthracis. BMC Microbiol. 2001;1:2.
9. Chang CH, Chang YC, Underwood A, Chiou CS, Kao CY. VNTRDB: a bacterial variable number tandem repeat locus database. Nucleic Acids Res. 2007 Ene;35 (Database issue):D416-21
10. Benson G. Tandem repeats finder: a program to analyzed DNA sequences. Nucleic Acids Res. 1999;27:573-580.
11. Kolpakov R, Bana G, Kucherov G. mreps: efficient and flexible detection of tandem repeats in DNA. Nucleic Acids Res. 2003;31:3672-3678.
12. Wexler Y, Yakhini Z, Kashi Y, Geiger D. Finding approximate tandem repeats in genomic sequences. J. Comp. Biol. 2005;12:928-942.
13. Denoeud F, Vergnaud G. Identification of polymorphic tandem repeats by direct comparison of genome sequence from different bacterial strains: a Web-based resource. BMC Bioinformatics. 2004;5:4.
Recibido: 25 de mayo de 2013.
Aprobado: 5 de junio de 2013.


{kind=link}