










My SciELO
 Custom services
Custom servicesServices on Demand
Journal

Article
 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista Cubana de Informática Médica
On-line version ISSN 1684-1859
RCIM vol.11 no.1 Ciudad de la Habana Jan.-June 2019 Epub June 01, 2019
Artículo original
Metodología para el minado in silico de loci polimórficos en microsatélites
1 Departamento de Bioquímica, Universidad de Ciencias Médicas, ICPB “Victoria de Girón”, La Habana, Cuba
Los polimorfismos con número variable de repeticiones en tándem (VNTR), constituyen marcadores genéticos utilizados en áreas de la genómica como estudios evolutivos, epidemiológicos y de genética poblacional. Los bancos de secuencias genómicas y las herramientas computacionales como BLAST permiten el minado de estos marcadores sin utilizar métodos experimentales, extendiéndolo a organismos no modelos de importancia médica o económica. Debido a la baja complejidad de estas secuencias y el número de candidatos que se presentan al inspeccionar un genoma cuando el procedimiento es escalado, surgen dificultades para procesar el volumen de datos generado y detectar por inspección visual los polimorfismos en los marcadores candidatos.
Se presentan una metodología y varios software que permiten la identificación y extracción rápida y fiable de loci polimórficos de SSRs. El procesamiento se hace por la concatenación de los programas MIDAS, BLAST, y el script PSSR-Extractor. Las entradas son rutas de directorios donde se encuentren múltiples archivos de secuencia en formato FASTA o GBFF y las salidas son los SSRs, códigos de acceso al GenBank, posiciones en el genoma, número de repeticiones y el grado de polimorfismo expresado como rango de variación, frecuencia alélica, cantidad de alelos y contenido de información polimórfica (PIC). Un script opcional, SSRMerge, permite la identificación de loci únicos (no redundantes) a nivel de especie, de género o en general del conjunto las secuencias que se desee procesar.
Se procesaron 23 genomas completos (RefSeq del NCBI) pertenecientes a diversos aislamientos de Mycobacterium tuberculosis. Se detectaron 4433 SSRs extrayéndose 414 loci no redundantes dentro de la especie. Realizado el minado de polimorfismos en las salidas del servidor BLAST para estos SSRs se reportan medidas que reflejan las variaciones que presentan estos loci.
Palabras Clave: SSR; VNTR; marcador molecular; minería de datos; algoritmo
Introducción
Los microsatélites, o repeticiones de secuencia simple (SSR, por sus siglas en inglés), son pequeños motivos de ADN (entre 1 y 6 nucleótidos) repetidos en tándem, presentes en todos los genomas de organismos procariotas y eucariotas 1. Estas secuencias se encuentran formando tractos que pueden ir desde unas pocas copias hasta cientos de ellas. El mecanismo molecular que explica estas secuencias es el llamado deslizamiento de la replicación, siendo el propio mecanismo el causante de la variabilidad observada en el número de copias 2. Los microsatélites presentan altos niveles de polimorfismo, que se traduce en el número variable de repetidos en tándem (VNTR, siglas en inglés) 3, con tasas de mutación entre 10-2 y 10-5 por locus por generación, contrastando con las de otros valiosos marcadores, por ejemplo los polimorfismos de simple nucleótido (SNP) que presentan una tasa de mutación alrededor de 10-9. La variación en las tasas de mutación de los VNTR produce igualmente un amplio rango de diversidad alélica haciendo a estos marcadores muy valiosos para determinar el grado de relación biológica entre poblaciones de una misma especie.
Cuando se escogen los microsatélites como marcadores para determinado estudio, los investigadores tienen dos opciones para su detección y caracterización: generar datos de secuencia o hacer minería en repositorios de secuencias ya sean públicos o privados. La primera opción requiere la preparación de librerías genómicas, plataformas de secuenciación y software para la detección posterior. La segunda opción hace los dos primeros pasos de la primera opción innecesarios, eliminando sus elevados costos, quedando solo la etapa asistida por software 4.
El minado in silico de SSRs polimórficos comprende dos etapas: 1ra detectar los SSR, para la cual se han desarrollado una amplia gama de aplicaciones, que, a pesar de tener el mismo fin, emplean criterios estadísticos y computacionales diversos los cuales influyen en los resultados que se obtienen 5,6; 2da determinar si estos SSR presentan polimorfismos en el número de copias, para lo cual es necesario hacer una comparación de sus secuencias contra repositorios de secuencias de especies relacionadas o de la misma especie. Las aplicaciones ideales para este fin son las de tipo BLAST (Basic Local Alignment Search Tool, http://blast.ncbi.nlm.nih.gov/) 7. Esta segunda etapa, debido al propio objetivo del BLAST de buscar mediante alineamiento local subsecuencias homólogas y a la naturaleza propia de los SSR, que son regiones de muy baja complejidad, conlleva un post-procesamiento de las salidas que presenta serias dificultades cuando queremos hacer esta minería a gran escala. Este post-procesamiento se hace normalmente editando manualmente los alineamientos e inspeccionándolos visualmente cuando son pocos los SSR candidatos a explorar, pero cuando se trata de cientos o miles de candidatos, requiere necesariamente una formalización y automatización del proceso. En artículos revisados sobre el tema, la metodología para estos fines no está esclarecida, presentándose en algunos casos de manera poco explícita y en otros omitiéndose completamente. El hecho de que aparezcan pocas referencias sobre esta segunda etapa se debe a que el procedimiento habitual es pasar directamente al genotipado funcional de forma experimental, para lo cual se requiere un extenso y costoso sistema de detección que incluye PCR, electroforesis y en ocasiones secuenciación.
En el caso de los microsatélites, la determinación del polimorfismo por variación en el número de copia, ya sea experimentalmente o in silico, se soporta en la especificidad de los flancos en cada locus. Estas secuencias flancos 5´ y 3´, que normalmente se reportan por los software que detectan SSR con tamaños alrededor de 20pb, se asumen conservadas y únicas en los genomas de cada especie, permitiendo la ubicación no ambigua del locus y convirtiéndose en las candidatas a secuencias cebadoras para la técnica de PCR.
Cuando hacemos una búsqueda extensiva con BLAST (blastn, para nucleótidos) y las secuencias de consulta (query) son SSR con sus respectivos flancos 5´ y 3´, se nos presentan varias complicaciones debido justamente a la baja complejidad de dichas secuencias y a la posibilidad de que los flancos no estén debidamente conservados. Debemos recordar que el BLAST es un sistema diseñado para detectar secuencias homólogas, y precisamente incluye filtros para de alguna forma eliminar las elevadas puntuaciones (score) que producen estas regiones repetidas. Este sistema no tiene un diseño específico para detectar secuencias homólogas no redundantes separadas por una región redundante y así captar el locus completo y poder comparar las variaciones en el número de copias.
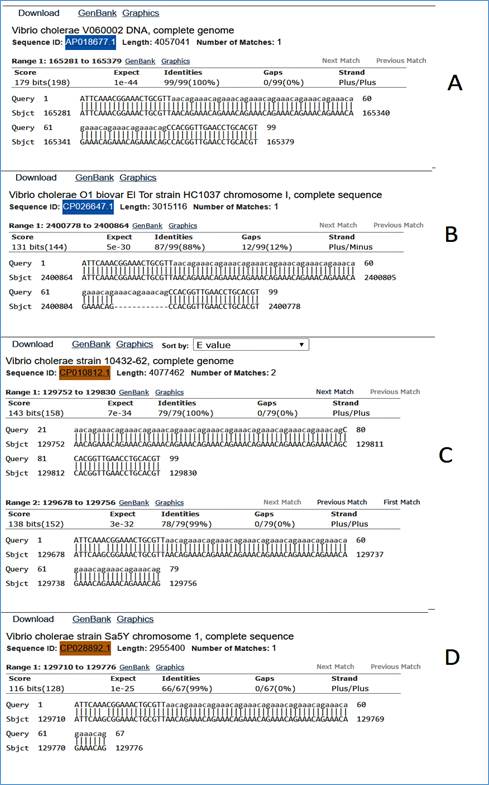
En las Figuras 1A-1D se observan las salidas BLAST para un SSR con dichas características y los distintos tipos de alineamientos que producen los hallazgos (hits) para diferentes entradas en la base de datos escogida. Se trata de un SSR extraído de un genoma de vibrio cholerae enfrentado a una base de datos de secuencias nucleotídicas de esta especie bacteriana. El SSR query se muestra con flacos de 20 bp en letras mayúsculas y la región repetida, que es la región que debe variar en longitud en caso de ser un locus polimórfico, en letras minúsculas, con motivo de repetición aacaga. En la Figura 1A se observa como el blastn encuentra una secuencia que es idéntica al query, produciendo un 100% de identidad y un e-value de 1e-44. En este caso no se descarta que la secuencia encontrada (ID: AP018677.1) sea la misma de la cual se extrajo el SSR aunque no necesariamente tiene que ser así. En Fig. 1B se observa un caso ideal donde se encuentra una entrada en la base de datos (ID: CP026647.1) con un locus que presenta variación en el número de repeticiones, en este caso es una supresión de (aacaga)2. En la Fig. 1C se observan dos hits para una misma secuencia (ID: CP010812.1), que representan dos entradas en el fichero de salida del BLAST (hit-table) donde no se pudo cubrir el locus completamente por el sistema y se presentan dos alineamientos que se solapan en determinada región. Este representa un caso donde es complicado detectar, visualmente o por otro tipo de método, la variación en el número de copias. Por último, en Fig. 1D se observa una entrada para una secuencia (ID: CP028892.1) donde el alineamiento se truncó, no llegando hasta el otro flanco y no reportando ningún otro hit para esa misma secuencia que contuviera el flanco derecho.
Estos son algunos ejemplos donde se observan las complicaciones que pudiera presentar, para un marcador, el interpretar y detectar el polimorfismo cuando se hace computacionalmente. Cuando esto es escalado a cientos de marcadores es totalmente imposible hacerlo por simple inspección visual de los alineamientos, incluso editando estos para resolver las entradas truncadas.
La metodología que presentamos describe las etapas y las bases algóritmicas para la detección computacional de polimorfismos de SSRs. El procedimiento general se hace por la concatenación de software que van desde la detección de los SSR, el procesamiento de los mismos por el sistema BLAST y la interpretación de las salidas del BLAST para la detección de los marcadores polimórficos.
En la siguiente sección (Métodos) se describe en detalle la secuencia de pasos que sigue esta metodología y los softwares empleados con la explicación de sus especificidades. En la sección Resultados, se expone y analiza la salida correspondiente a la detección de polimorfismos de SSRs en genomas de Mycobacterium tuberculosis. También se describen los parámetros de entrada, formatos de entrada y salida y los valores reportados.
Métodos
En la Figura 2 se presenta la secuencia general de etapas para el minado in silico de SSRs polimórficos. Primeramente se hace una corrida de MIDAS 8 entrándole como parámetros un fichero de secuencia genómica que puede ser en formato FASTA o GBFF (de entradas sencillas o múltiples), la unidad mínima del repetido a detectar y los parámetros del alineamiento para match, mismatch e indel. MIDAS detecta todos los SSRs, exactos o aproximados, y genera un fichero MultiFASTA (extensión .mfaa) con ellos a los que le añade secuencias flancos de 20 bp en letras mayúsculas y la secuencia repetida en letras minúsculas. Este formato que marca la región repetida con letras minúsculas es utilizado por el BLAST como forma de enmascaramiento.
Seguidamente se corre el script SSRMerge que permite extraer el conjunto de SSR no redundantes a partir de múltiples ficheros de salida del MIDAS aplicados a múltiples genomas. Su primer parámetro es un camino en el directorio de la PC y procesará todos los ficheros con extensión .mfaa que encuentre en su interior. Esta etapa es opcional y es aplicable solo cuando estamos analizando múltiples genomas de especies relacionadas. Mientras mayor sea el vínculo taxonómico entre estos genomas, mayor será la probabilidad de encontrar locus similares conservados que se repiten en distintos genomas. El principio algorítmico de este script se basa en una comparación de los flancos de los SSR, todos contra todos, presentes en todos los ficheros que se procesen. Esta comparación se hace por alineamiento de secuencia global Nedleman-Wunsch. Cuando al comparar dos SSR y estos presentan más de un 90% de identidad se escoge uno de ellos y se desecha el otro. El resultado es un fichero MultiFASTA que contiene un conjunto de SSR no redundantes, de acuerdo a los parámetros definidos, y asumimos que estos SSRs pertenecen a loci distintos dentro del conjunto de genomas analizados.

La tercera etapa es una corrida BLAST (NCBI server, http://blast.ncbi.nlm.nih.gov/). La entrada al servidor es el fichero MultiFASTA de la etapa previa. BLAST tiene muchos parámetros de configuración para hacer una corrida. Los parámetros que debemos modificar en nuestro caso particular son los siguientes: En el conjunto de búsqueda (Choose Search Set) especificar el organismo para el cual queremos hacer la búsqueda (e.g. Mycobacterium tuberculosis (taxid:1773)); en el tipo de programa seleccionar blastn, programa diseñado para encontrar homologías más remotas; en parámetros generales del algoritmo solo nos interesa modifica el umbral esperado (expect threshold) que por defecto es 10. El umbral esperado debe ser incrementado a >30 permitiéndonos encontrar hits que aunque tengan e-values grandes pueden ser loci de interés y la etapa siguiente ejecutada por PSSRextractor se encarga de depurar éstos. Otro parámetro que es necesario cambiar es el de enmascarado (mask). Aquí debemos marcar la opción de regiones de baja complejidad (low complexity regions) y enmascarar con letras minúsculas (mask lower case letters). Esto garantiza que el blastn intente reconocer todo el lucus incluyendo los dos flancos, aunque esto no siempre se logra, sobre todo cuando la región repetida es grande. El script PSSRextractor tiene implementadas reglas de negocio para solventar estos casos. La salida de esta etapa es el fichero texto de tabla de éxitos (hit-table) que genera el propio servidor.
Por último, la salida del BLAST es procesada por PSSRextractor. Este puede procesar uno o varias salidas pues su primer parámetro de entrada es un camino a un directorio, y procesará todos los ficheros de salidas BLAST que encuentre en su interior. Este script primeramente analiza sintácticamente (parser) la salida hit-table extrayendo toda su información. Los otros dos parámetros del script son porciento de identidad y porciento de cubrimiento que definen los criterios para tener en cuenta los flancos a considerar en cada entrada de la hit-table. Posteriormente procede a evaluar los polimorfismos por número variable de repeticiones presente en estos datos y lo hace de la manera siguiente:
Para un SSR (query en hit-table) habrá una o muchas entradas (subject en hit-table). Cada SSR tiene un tamaño de la unidad repetida (RUS). Cada entrada en hit-table tiene, entre otros, los siguientes valores: identificador de acceso del subject (SAV), longitud del alineamiento (AL), % de identidad (%I), posición inicial del query (q.start), posición final del query (q.end), posición inicial del subject (s.start) y posición final del subject (s.end). Entonces con estos datos se calcula el número de repeticiones (RN) de cada unidad repetida validando las siguientes condiciones:
Si AL > 40 Entonces: RN = (AL - 40) / RUS. Cuando el alineamiento es mayor o igual a 40 significa que cubrió ambos flancos del query, dado que la región repetida está marcada y no se tiene en cuenta, entonces la diferencia nos daría la cantidad de nucleótidos que están en la región repetida que dividida entre RUS nos devuelve el RN. Estos casos ocurren con muy baja frecuencia.
Si q.end <= 20 Entonces: La secuencia encontrada coincide con el flanco izquierdo. De lo contrario: La secuencia encontrada coincide con el flanco derecho. De modo que para un mismo query, todas las entradas con el mismo SAV, se clasificaran a la izquierda o a la derecha del locus según esta condición. Estos casos son los que ocurren con mucha frecuencia. Ver Figura 3.
Si s.start izquierda < s.end izquierda && s.start derecha < s.end derecha && s.end izquierda < s.start derecha Entonces: RN = |s.start derecha - s.end izquierda| - 1 / RUS. Esta condición valida que el flanco izquierdo quede a la izquierda de la secuencia subject y que el flanco derecho está a la derecha, siendo la positiva dirección (es decir de 5´ a 3´). Similar condición se valida cuando la dirección es negativa solo hay que invertir las desigualdades estrictas. Esto ocurre porque el BLAST analiza también la complementaria de la secuencia en la BD (Fig. 3 y 4).
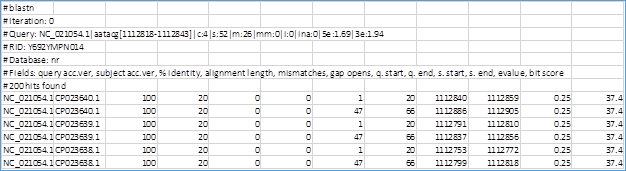
Fig. 3 Ejemplo de salida hit-table del BLAST. Se observa como para un query encuentra dos entradas para un mismo subject con SAV CP023640.1. La primera entrada tiene q.start=1 y q.end=20 (flanco izquierdo del query) y la segunda tiene q.start=47 y q.end=66 (flanco derecho del query).

Fig. 4- Validación de la dirección (positiva o negativa de la comparación query-subject) y cálculo de RN.
RN escogido = min {RNs encontrados}. Esta validación se realiza porque se pueden dar casos dudosos debido a duplicaciones de regiones en un mismo genoma. Por ejemplo, para el query con la unidad repetida AATACG (zona roja) entre el flanco izquierdo (zona verde) y flanco derecho (zona azul) se pueden encontrar los siguientes casos (Fig.5):

Solo se tienen en cuenta entradas donde los alineamientos tengan un %I (segundo parámetro del script) y un % de cubrimiento (tercer parámetro del script, (AL/20) x 100) mayores que 90. Estos dos parámetros garantizan que los flancos encontrados estén bien conservados.
Luego de tener el conjunto de NR para todos los subjects de un query, PSSRextractor genera dos ficheros de resultados, uno detallado y otro genérico, con nombres iguales al de la hit-table pero con los sufijos _specific.xls y _generic.xls respectivamente. El informe detallado brinda información sobre cada subject procesado y el genérico brinda la información relacionada al polimorfismo para cada query, es decir para cada SSR.
Entre la información relacionada al polimorfismo que brinda el reporte genérico se encuentran los siguientes valores:
min_RN, max_RN y range: Son tres columnas en el reporte que significan respectivamente el RN mínimo, el RN máximo y el rango (max_RN - min_RN).
frecuency: Frecuencia del alelo (número de repeticiones de la unidad repetida, NR, entre el total de todos los alelos) que presenta el SSR original a partir del cual se hizo la búsqueda.
alleles: Número de alelos encontrados para un locus (SSRs con RN diferentes).
PIC: Contenido de Información Polimórfica
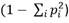 . Este valor también se conoce en otros contextos como heterocigocidad promedio esperada o diversidad genética de Nei, y da una medida de la probabilidad de que, para un locus único, un par de alelos escogidos al azar en la población sean diferentes.
. Este valor también se conoce en otros contextos como heterocigocidad promedio esperada o diversidad genética de Nei, y da una medida de la probabilidad de que, para un locus único, un par de alelos escogidos al azar en la población sean diferentes.
El resto de los valores que muestra el reporte genérico provienen de MIDAS (access_number, pattern, pattern_length, RN, inaccuracy, entropy_5, entropy_3), la cual aporta información valiosa al interpretar los SSR polimórficos. Por ejemplo el grado de inexactitud encontrado en el SSR o la entropía composicional de los flancos, nos permiten conocer respectivamente el grado de inexactitud del tracto repetido y cuán informativos pueden ser los flancos en la caracterización del locus.
Los valores de la última columna del reporte genérico (exceptions) muestran etiquetas que corresponden a excepciones en las validaciones del polimorfismo. Hay entradas en el reporte genérico donde pueden aparecer más de una de estas etiquetas pues las excepciones se pueden dar simultáneas. Si todas las secuencias subject presentan excepciones, ya sea de unos o de otras, entonces se colocan las etiquetas. Las etiquetas son las siguientes:
D (degenerated): Los subjects tiene un %I y/o un % cubrimiento < 90%.
NF (not found): No se encontró ningún subject en la base de datos con similitud.
O (outlier): La cantidad de unidades repetidas entre los flancos de los subjects es dudosa por ser muy grande, siendo improbable que exista un microsatélite entre ellos. Los valores de corte establecidos para establecer esta excepción fueron mononucleótido: 157 bp, dinucleótido: 364 bp, trinucleótido: 109 bp, tetranucleótido: 45 bp, pentanucleótido: 150 bp y hexanucleótido: 193 bp. Estos valores fueron definidos después de procesar todos los SSR de más de 200 genomas bacterianos, registrando sus tamaños, y estableciendo el corte en 3 veces el rango intercuartil.
U (unpair): Para una misma secuencia subject aparece un flanco y no el otro.
Resultados y Discusión
Mycobacterium tuberculosis (conocido también como bacilo de Koch) es un patógeno bacteriano, agente causal de la tuberculosis, infección contagiosa que afecta principalmente a los pulmones pero puede propagarse a otros órganos. La prevalencia de esta enfermedad es muy alta a nivel mundial, existiendo aproximadamente 12 millones de personas infectadas. El bacilo es también objeto de preocupación dentro de la comunidad médica y científica por presentar cepas con resistencia a múltiples antibióticos.
Los errores por deslizamiento en la replicación causantes de los microsatélites son reparados normalmente por tres enzimas mutL, mutS y mutH, sin embargo algunos genomas como los de micobacterias adolecen de este sistema enzimático 9. Debido a ello estas especies bacterianas constituyen un valioso ejemplar para investigar las tasas de mutación de microsatélites y sus mecanismos regulatorios 10.
M. tuberculosis es un patógeno con una diversidad genética emergida de cepas más diversas, ganando así en sus mecanismos de virulencia 11. Un ejemplo de importancia médica es la expansión de un microsatélite de micobacterias que ocurre en proteínas con el pentapétido-2 (PP2) 12.
En el presente estudio se analizaron 23 genomas completos (secuencias RefSeq), descargados del sitio ftp del NCBI (ftp://ftp.ncbi.nlm.nih.gov/refseq/release/bacteria/), pertenecientes a diversos aislamientos de M. tuberculosis. MIDAS detectó 4433 SSRs y de ellos se extrajeron con el script SSRMerge 414 loci no redundantes dentro de la especie. Se hizo el minado de polimorfismos para las salidas del servidor BLAST para estos SSRs con el script PSSRextractor. Las salidas de estos dos scripts pueden consultarse en los ficheros adjuntos (species_all.mfaa y Y692YMPN014-Alignment_generic_result.xls).
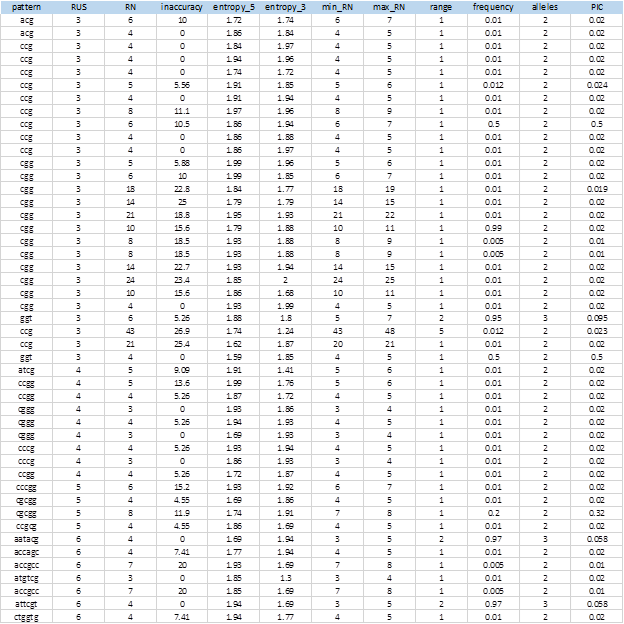
De los 414 SSR, fueron elegidos 288 que no mostraron ningún tipo de excepción, y de éstos, 104 mostraron PIC > 0, (36,1%). La Figura 6 muestra los valores promedios obtenidos para los diferentes tipos de SSRs clasificados por el tamaño de la unidad repetida (RUS).

Fig. 6 Valores promedios para algunas medidas extraídas del reporte genérico en la especie M. tuberculosis.
Es significativa la cantidad de SSRs con 3bp como unidad repetida (76%). Esto es debido a que la mayor parte de los genomas bacterianos son regiones codificantes, las cuales presentan sesgo composicional en sus codones. Se detectó un único SSR para dinucleótidos y fue excluido del resultado por presentar excepciones en el análisis de polimorfismo. Como promedio, los SSR de 1bp fueron más inexactos y mostraron mayor polimorfismo con más cantidad de alelos y un PIC elevado (3.17 y 0.08 respectivamente). Los tractos de repeticiones (RN) fueron también significativamente mayores al resto.

En la Figura 7 se muestra el listado completo de los 104 SSR extraídos que mostraron algún nivel de polimorfismo (PIC > 0). Las secuencias íntegras de los marcadores, incluidas las secuencias flancos, los números de acceso y las posiciones en el genoma pueden ser obtenidas de las salida de MIDAS.
La metodología descrita tiene aspectos distintivos con respecto a otros procedimientos in silico reportados en la literatura:
Se ha demostrado en ensayos experimentalmente que no todos los loci de SSRs muestran polimorfismo. Esto se debe, entre otros aspectos, a que el locus dentro de la población analizada no se encuentra sujeto a una dinámica de cambio en particular. En este sentido, la metodología nos permite seleccionar aquellos loci que sí manifiestan polimorfismos en los bancos de secuencia escogidos, reduciendo los costos que esto implica cuando se hace de forma experimental.
La determinación del polimorfismo presente en el locus es totalmente automatizada. Los procedimientos comunes, no experimentales, emplean el alineamiento múltiple de los marcadores para luego, por inspección visual, detectar los polimorfismos. En este sentido, el procedimiento es ideal para el análisis a gran escala.
El polimorfismo se define estrictamente como variación en el número de copias del SSR (PIC > 0), y no como simples inserciones o supresiones presentes en los marcadores que no correspondan con el tamaño de la unidad repetida.
El procedimiento puede hacerse partiendo de la detección de SSRs en una secuencia, pero opcionalmente, también puede partir de múltiples secuencias sin necesidad de hacer ensamblaje para obtener una secuencia consenso. Esto permite detectar SSRs que no pertenecen al mismo locus a pesar de estar en genomas muy emparentados. El script SSRMerge permite eliminar la redundancia de los loci comunes a todas las secuencias.
La metodología es excelente para el análisis puramente computacional de loci de SSRs, en estudios evolutivos, de identificación genotípica o estudios funcionales a partir de los genes involucrados. Para su uso en el análisis experimental utilizando PCR, la metodología brinda toda la información necesaria (identificador de acceso a la secuencia, posiciones en el genoma, secuencias flancos, etc.) que permite el diseño de cebadores utilizando otras aplicaciones disponibles en internet.
Disponibilidad
Todos los software están disponibles en el material suplementario: MIDAS (distribución binaria midas_v1.1.exe), SSRMerge and PSSRExtractor se suministran en formato comprimido zip (ambos son Java NetBeans Projects para JDK 1.8 o superior, y los ficheros binarios jar, para ejecutar por línea de comandos, están en la carpeta \dist una vez descomprimidos).
Conclusiones
Se describe una metodología para la detección totalmente computacional de loci polimórficos de microsatélites. Como ejemplo de su utilización se procesaron 23 genomas completos pertenecientes a diversos aislamientos de M. tuberculosis. Se detectaron 4433 SSRs y de ellos se extrajeron 414 loci no redundantes dentro de la especie. Se hizo el minado de polimorfismos para las salidas de BLAST y 100 SSRs mostraron polimorfismos. La metodología es intuitiva y viene acompañada de software para su aplicación. Su principal ventaja radica en los niveles de escalado que permite y la reducción de costos cuando se hacen análisis experimentales permitiendo la preselección de marcadores que han evidenciado polimorfismo en bancos de secuencias genómicas escogidos.
References
1. Li YC, Korol AB, Fahima T, Beiles A, Nevo E. Microsatellites: Genomic distribution, putative functions and mutational mechanisms: A review. Molecular Ecology 2002; 11: 2453-2465. [ Links ]
2. Ellegren, H. Microsatellites: Simple sequences with complex evolution. Nature Reviews. Genetics 2004; 5: 435-445. [ Links ]
3. Xu, J.S.,Wu,Y.T.,Ye,S.J.,Wang,L.,and Feng,Y.Z. SSR primer screening and assessment on pear germplasm resources. J. Central South Univ. Forest.Technol. 2012; 32, 80-85. [ Links ]
4. Hodel et al. Using microsatellites in the 21st century. Applications in Plant Sciences 2016 4(6) [ Links ]
5. Leclercq, S., Rivals, E., Jarne, P. Detecting microsatellites within genomes: significant variation among algorithms. BMC Bioinformatics 2007, 8:125. [ Links ]
6. Grover A, Aishwarya V, Sharma PC. Searching microsatellites in DNA sequences: approaches used and tools developed. Physiol Mol Biol Plants (January-March 2012) 18(1):11-19 [ Links ]
7. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J. Mol. Biol. (1990) 215:403-410. [ Links ]
8. Martínez CM. MIDAS: Computer application for the identification of exact and inaccurate microsatellites in genomic sequences. Revista Cubana de Informática Médica, Volúmen 18, No. 2 (2018). [ Links ]
9. Fleischmann RD, Alland D, Eisen JA, Carpenter L, White O, Peterson J, DeBoy R, Dodson R, Gwinn M, Haft D, Hickey E, Kolonay JF, Nelson WC, Umayam LA, Ermolaeva M, Salzberg SL, Delcher A, Utterback T, Weidman J, Khouri H, Gill J, Mikula A, Bishai W, Jacobs Jr WR, Venter JC, Fraser CM: Whole-genome comparison of Mycobacterium tuberculosis clinical and laboratory strains. J Bacteriol 2002, 184(19):5479-5490. [ Links ]
10. Sreenu V, Kumar P, Nagaraju J, Nagarajaram H. Microsatellite polymorphism across the M. tuberculosis and M. bovis genomes: Implications on genome evolution and plasticity. BMC Genomics 2006, 7:78. [ Links ]
11. Supply P, Marceau M, Mangenot S, Roche D, Rouanet C, Khanna V, et al. Genomic analysis of smooth tubercle bacilli provides insights into ancestry and pathoadaptation of Mycobacterium tuberculosis. Nat Genet. 2013 Feb; 45(2):172-179. doi: 10.1038/ng.2517 PMID: 23291586. [ Links ]
12. Warholm P, Light S. Identification of a Non-Pentapeptide Region Associated with Rapid Mycobacterial Evolution. PLoS ONE (2016), 11(5): e0154059. doi:10.1371/journal.pone.0154059 [ Links ]
Recibido: 20 de Abril de 2018; Aprobado: 24 de Mayo de 2018
 This is an open-access article distributed under the terms of the Creative Commons Attribution License
This is an open-access article distributed under the terms of the Creative Commons Attribution License
