










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkMediSur
versión On-line ISSN 1727-897X
MediSur v.7 n.3 Cienfuegos Mayo-jun. 2009
Folletos
Revista Científica de las Ciencias Médicas en Cienfuegos
El ABC para el diagnóstico y prevención de las enfermedades genéticas en la atención primaria de salud.
Julia Surí González,1 María Antonia Ocaña Gil,2 María del Rosario Liriano Ricabal,3 Luisa Días Requeiro,4 Antonio Masót Rangel,5 Sonia González Sosa,6
Resumen
Los programas para la prevención de las enfermedades genéticas logran reducir las nefastas consecuencias que estas producen en la población. Por ello, es fundamental que la preparación del personal que de una forma u otra está vinculado al Programa Materno-Infantil, sea lo más completa y actualizada posible. La docencia relacionada con estos temas se imparte desde el pregrado, en el segundo año de la carrera de Medicina, y también en la enseñanza posgraduada, en el segundo año de la residencia en Medicina General Integral, a través de la asignatura Genética Médica. La importancia que reviste este tema sirvió de fundamento para la confección del presente folleto, que tuvo como punto de partida una revisión bibliográfica sobre los temas correspondientes a la aplicación del programa de diagnóstico y prevención de enfermedades genéticas, en el contexto de la práctica del asesoramiento genético. El objetivo de este folleto es ofrecer los elementos teóricos básicos, necesarios para facilitar las acciones de salud en la comunidad, que pueden ser posteriormente profundizados mediante la revisión de la bibliografía convencional.
Palabras clave: Enfermedades genéticas congénitas;genética medica;servicios genéticos;diagnostico;prevención de enfermedades;
INTRODUCCIÓN
Registros estadísticos realizados por la Organización Mundial de la Salud (OMS), informan que de un 1,5-6,5 % de los recién nacidos tiene una anomalía congénita de diferente tipo y que cerca de un 2-3 % nace con severas y hasta letales anomalías genéticas, lo que en nuestro país corresponde a una tasa de 3,8-3,9 por 1 000 nacidos vivos.
En Cuba, se calcula que alrededor de 2 000 niños al año nacen con algún tipo de enfermedad hereditaria y que un elevado por ciento de estas se acompaña de alteraciones neurológicas graves, las cuales pueden llevar a la muerte o a situaciones de discapacidad.
Es obvio que llevar a cabo un programa para la prevención de las enfermedades genéticas logra reducir las nefastas consecuencias que estas producen en la población; dentro de este programa ocupa primordial valor la pesquisa para el diagnóstico y prevención de las malformaciones congénitas y enfermedades hereditarias.
Para llevar a cabo esta labor es importante la adopción y cumplimiento del programa en la Atención Primaria de Salud y más específicamente en los Consultorios Médicos de la Familia, de ahí la importancia de contar con un manual que recoja sus principales aspectos, de forma tal que pueda ser útil no sólo para los médicos y enfermeras de la familia, sino también para todos aquellos que de una forma u otra estén relacionados con el noble empeño de proporcionar a la pareja la posibilidad de tener hijos sanos, y en una dimensión más amplia desde el punto de vista social, disminuir los índices de discapacidad y elevar la calidad de vida del pueblo.
Por ello, el objetivo del presente folleto es ofrecer los elementos teóricos básicos necesarios para facilitar las acciones de salud en la comunidad, los cuales pueden ser posteriormente profundizados en la bibliografía convencional.
CAPÍTULO I. ACCIONES DE SALUD EN LA ATENCIÓN PRIMARIA PARA LA PREVENCIÓN DE ENFERMEDADES GENÉTICAS.
La Atención Primaria de Salud es el escenario fundamental para el control y seguimiento a las embarazadas, esencial para el desarrollo del programa de diagnóstico y prevención de las enfermedades genéticas. Es aquí donde se le indican a la embarazada una serie de estudios (Cuadro 1):
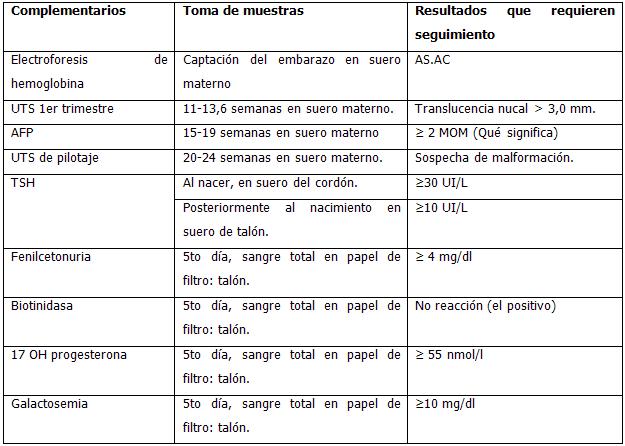
- Electroforesis de hemoglobina a la captación del embarazo. (Gráfico 1)
- Valoración del riesgo genético por el máster en Asesoramiento Genético (AG) en Consulta Multidisciplinaria del área de salud una vez realizada la captación del embarazo y orientación de seguimiento a toda gestante según el riesgo.
- Ultrasonido (UTS) en el primer trimestre entre las 11 y 13,6 semanas.
- Alfafetoproteína (AFP) entre las semanas 15 y 19 de gestación. (Gráfico 2)
- Ultrasonido para la detección de malformaciones congénitas (pilotaje) entre las 20 y 24 semanas de gestación (preferiblemente entre las 22 y 24 semanas).
- Seguimiento por interconsulta a toda gestante con alteraciones en las pruebas de pesquisa correspondientes: hemoglobinopatía, alfafetoproteína, diagnóstico prenatal cromosómico (DPC) (Gráfico 3), ultrasonido y ecografía fetal.


Una vez que nace el niño, se indican otros exámenes:
- Cuantificación de hormona tiroestimulante (TSH) en muestra de sangre del cordón umbilical. Recordar, en caso de un parto extrahospitalario, con las condiciones mínimas para realizar este estudio, que son suficientes sólo 5 cc de sangre de la porción del cordón correspondiente al feto, pero sin ordeñarlo. (Gráfico 4)
- A los 5 días de nacido se realiza la pesquisa neonatal en muestras de sangre de talón (Gráfico 5):
- Fenilcetonuria.
- Hiperplasia adrenal congénita.
- Déficit de biotinidasa.
- Galactosemia.
3. Evaluación de todo recién nacido por el máster en AG de la comunidad y seguimiento (si hay riesgo genético incrementado).

Si se detectan entidades de etiología genética al nacimiento o en cualquier etapa de la vida deben ser registrados y ofrecérsele atención multidisciplinaria (evaluación, estudio y seguimiento por máster en AG y otras especialidades según corresponda).
Es importante tener en cuenta que todo paciente que presente riesgo genético incrementado o solicite ser asesorado desde el punto de vista genético debe recibir este servicio por el máster en AG en la atención primaria de salud a través de las consultas de:
- Riesgo genético preconcepcional.
- Riesgo genético prenatal.
- Riesgo genético pediátrico.
- Riesgo genético del adulto.
CAPÍTULO II. PESQUISA PRENATAL.
Diagnóstico prenatal de anemia por hematíes falciformes.
La enfermedad genética más frecuente en Cuba es la anemia falciforme, conocida también como siclemia. Para detectarla existe un programa que se ocupa de las embarazadas portadoras de esta enfermedad.
La enfermedad se produce por una mutación en la posición 6 del gen betaglobina, en la cual el aminoácido valina sustituye al ácido glutámico presente en la hemoglobina A (hemoglobina normal), originando la hemoglobina S (hemoglobina anormal). La hemoglobina S, en condiciones de baja tensión de oxígeno, es menos soluble que la hemoglobina A y forma agregados al unirse entre sí, lo cual afecta la capacidad de transportación del oxígeno y deforma la estructura de la membrana citoplasmática, normalmente muy flexible. Este hematíe deformado es el causante de toda la sintomatología y complicación de la enfermedad.
Las características clínicas más frecuentes son anemia severa, artralgias, dolores abdominales y óseos, cuyas manifestaciones son variables de un individuo a otro y evolucionan en forma de crisis (por episodios).
Se hereda de forma autosómica recesiva (AR).
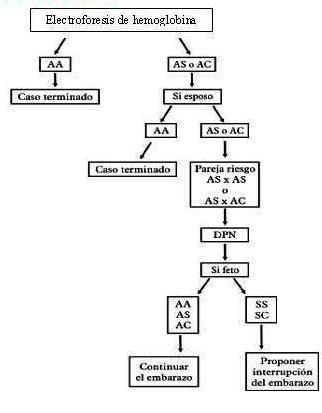
El mecanismo de prevención de esta enfermedad se basa en la pesquisa que se realiza en toda embarazada a través de la realización de electroforesis de hemoglobina, indicada por el médico de familia en la captación del embarazo. Sus resultados no están en relación con la edad gestacional, realizar esta prueba tempranamente ayuda en caso de detectar una pareja riesgo a realizar el DPN y a un adecuado AG.
Conducta según el resultado de la electroforesis de hemoglobina:
- Si la hemoglobina de la embarazada es AA (hemoglobina normal) no tiene riesgo de que su descendencia se vea afectada por esta enfermedad.
- Si la hemoglobina de la embarazada es AS o AC (hemoglobina anormal) se repite el estudio para confirmar el tipo de hemoglobina y se le indica al esposo.
- Si se confirma lo anterior (embarazada AS o AC) y el esposo es AA, no hay riesgo de descendencia afectada. Si el esposo es AS o AC, constituyendo pareja AS x AS, o AS x AC, se considera pareja de riesgo. En estos casos el riesgo es de un 25 % o ¼ de probabilidad para cada gestación de tener un hijo enfermo.
- Si constituyen pareja de riesgo tienen la opción del estudio prenatal para conocer el genotipo del feto y decidir el curso del embarazo.
Cuando es conocido que en una familia hay síntomas de cualquier hemoglobinopatía debe indicarse el estudio (electroforesis de hemoglobina) a todos sus miembros en edad fértil. El diagnóstico preconcepcional garantiza a la pareja conocer el diagnóstico en etapas tempranas y permite un asesoramiento genético oportuno.
Determinación de alfafetoproteína en suero materno como marcador de malformación fetal.
La alfafetoproteína (AFP) es una glicoproteína producida normalmente por el feto humano. Es sintetizada en las primeras semanas por el saco vitelino y posteriormente por el hígado y tracto gastrointestinal; se encuentra en el líquido amniótico en embarazos normales y atraviesa las membranas fetales y la placenta hacia el suero materno.
La síntesis de la AFP está controlada por el gen recesivo Rafp; este gen está bajo la influencia de un gen represor tras el nacimiento. Le han sido atribuidas varias funciones a esta proteína fetal, entre las cuales están el factor de crecimiento fetal, la albúmina fetal, el transporte de estrógenos y la regulación de la respuesta inmunológica en el embarazo.
Los niveles normales de AFP en suero materno aumentan en relación con la edad gestacional (EG), por lo que es necesario precisar adecuadamente la EG de la embarazada. Si existe un error de cuenta o no se precisa en la indicación de este estudio la EG, o fecha de la última menstruación, por ejemplo, puede ocurrir que un valor elevado para una EG supuesta de 15 semanas sea normal si el ultrasonido indica una EG de 19 semanas.
Los valores de la AFP son expresados en múltiplos de la mediana (MOM) para la EG y, por tanto, un mismo valor en nanogramos por mililitro puede diferir en MOM según la EG que se considere. Se considera elevada la AFP con valores a partir de 2 MOM (Cuadro 2). Por todo lo anterior se destaca la importancia de determinar la EG en el primer trimestre del embarazo.
Niveles bajos de AFP pueden estar causados por:
- No embarazo.
- Huevo huero
- Aborto diferido.
- Embarazo molar.
- Síndrome de Down.
El médico de familia indica a todas las embarazadas la AFP entre las 15 y 19 semanas de gestación. Después de esta EG no tiene valor el estudio.
Cuando una embarazada presenta cifras elevadas de AFP en suero materno, es preciso:
- Verificar la EG para descartar un error de cálculo.
- Revisar la historia obstétrica de sangrado u otros síntomas de amenaza de aborto, así como enfermedades maternas.
- La embarazada se cita a consulta de UTS de su municipio donde se evalúa por el máster en AG.
- Si el UTS es normal (no sospecha de malformación), se le da el alta en consulta de AFP.
- Si se sospecha de malformación, se le da seguimiento por UTS en el Centro Provincial de Genética Médica hasta las 26 semanas.
- Cuando es diagnosticada una malformación por UTS se propone la interrupción del embarazo, lo cual se lleva a cabo si la pareja lo desea.
Detección prenatal de malformaciones congénitas por ultrasonido.
Aunque no todas las malformaciones congénitas son diagnosticadas prenatalmente, un porcentaje importante sí puede ser diagnosticado.
El mejor momento para realizar el UTS control, llamado "UTS de pilotaje" es el que se realiza a todas las embarazadas indicado por él médico de familia para el diagnóstico de malformaciones congénitas entre las 20-24 semanas, preferiblemente a las 24 semanas. En este tiempo la mayoría de los órganos son visibles por UTS y hay suficiente tiempo para brindarle una opción a la pareja en caso de detectarse una anomalía o malformación fetal.
Diagnóstico prenatal de cromosomopatías.
Las enfermedades de origen cromosómico influyen notablemente en la morbimortalidad perinatal y son causa importante de retraso mental y abortos espontáneos. Se calcula que alrededor del 0,5 % de los recién nacidos vivos presenta aberraciones cromosómicas. De estas, la más frecuente es el Síndrome de Down, producido por trisomía 21, cuya incidencia es de 1:600 recién nacidos vivos.
El diagnóstico prenatal citogenético (DPC) es el examen que permite valorar el número y la estructura de los cromosomas fetales. A través del estudio del líquido amniótico, vellosidades coriónicas o sangre fetal se puede realizar el cariotipo fetal y conocer la dotación cromosómica del feto.
Dada la imposibilidad actual de realizar el DPC masivamente a todas las embarazadas es necesario seleccionar las embarazadas con riesgo incrementado, considerando como principales indicaciones las siguientes:
- Avanzada edad de la madre.
- Uno de los progenitores portador de aberración estructural balanceada.
- Hijo anterior con una cromosomopatía diagnosticada o sospechada.
- Enfermedades ligadas al sexo.
La indicación más frecuente para el DPC la constituye la avanzada edad materna, esta condiciona un mayor riesgo para que ocurra un fallo en la segregación de los cromosomas en la ovogénesis y explica la mayor incidencia de hijos afectados con Síndrome de Down por trisomía 21. Se le propone este estudio a todas las gestantes a partir de los 35 años de edad dada la cobertura existente en estos momentos para el estudio.
Probabilidad de tener un feto afectado con síndrome de Down al nacimiento según edad materna:

El DPC se puede realizar mediante biopsia coriónica (10-12 semanas de gestación) o amniocentesis (15-18 semanas de gestación).
A pesar de que el DPC se considera una técnica invasiva, con bajo riesgo de complicación maternofetal (del 1 %), en dependencia del método utilizado y la experiencia en dicho método, debe indicarse cuando el riesgo de aparición de una aberración cromosómica es lo suficientemente importante como para desestimar las posibles complicaciones.
A partir de 1990 se ha ido mejorando la evaluación por ultrasonido de todo el período de gestación. En este acápite se hace referencia específicamente al ultrasonido en el primer trimestre del embarazo y el diagnóstico precoz de las cromosomopatías. Uno de los parámetros analizados en este sentido ha sido el diámetro anteroposterior (espesor) de la translucencia nucal (TN). La TN aumenta a lo largo de toda la gestación y se considera siempre anormal cuando es mayor de 3, 0 mm entre las 11-14 semanas de gestación. El riesgo de anomalías aumenta con el aumento de espesor de la TN, si es de 3-4 mm el riego es del 10 %, si es de 4-6 mm el riesgo es de 40 %, si es mayor de 6 mm el riesgo es de 80 %.
La medición de la TN debe hacerse entre las 11-13, 6 semanas de gestación, lo que se correlaciona con embriones cuya longitud embrionaria máxima (LEM) es entre 45 y 84 mm respectivamente. Idealmente la medición debe hacerse por vía transvaginal por personal adecuadamente adiestrado. Además de la pesquisa de Síndrome de Down, la TN aumentada ha permitido detectar otras cromosomopatías: trisomía 13, 18, Síndrome de Turner, Triple XXX, 47 XYY, Síndrome Klinefelter y otros más.
En estas afecciones, además de la TN aumentada, pueden coincidir otros signos ecográficos que permiten sospechar las anomalías tales como la ausencia del hueso nasal, menor longitud del maxilar, menor volumen placentario (normal 3 cm), escaso crecimiento fetal, atresia umbilical única, onfalocede (fisiológico hasta las 12 semanas) y megavejiga.
En el primer trimestre del embarazo, ante una TN aumentada es necesario realizar:
- Un examen detallado de la anatomía fetal y considerar la determinación del cariotipo fetal (DPC).
- Exámenes periódicos de seguimiento por ultrasonido detallado del cariotipo fetal.
- Una ecocardiografía fetal alrededor de las 20 semanas.
- Serología completa a la madre para descartar infecciones intrauterinas.
- Descartar síndromes genéticos.
La remisión de la TN mejora el pronóstico fetal, por el contrario el aumento progresivo agrava el pronóstico.
CAPÍTULO III. PESQUISA POSNATAL.
Diagnóstico neonatal de hipotiroidismo congénito
El hipotiroidismo puede ser clasificado, de acuerdo al momento de la lesión, en: hipotiroidismo congénito, que se presenta en la etapa neonatal, e hipotiroidismo adquirido con una etapa de funcionamiento normal de la glándula y un comienzo posterior en la vida, con cuadro clínico menos severos y sin secuelas mentales irreversibles, cuando aparece después del 3er. año de edad.
El hipotiroidismo congénito (HC) está dado por una producción insuficiente de la hormona tiroidea desde los primeros tiempos de vida y constituye la causa más frecuente de retraso mental prevenible en la infancia. La prevalencia es cada día más uniforme a nivel mundial, se señala 1 caso por cada 3 000-4 000 recién nacidos. Dentro de su diagnóstico precoz hay que considerar, en especial, al causado por aplasia, hipoplasia o localización anómala del tiroides.
El cuadro clínico está en relación con el grado de insuficiencia tiroidea, la época de aparición, y el tiempo de evolución de la enfermedad sin tratamiento. Los síntomas que se exponen a continuación son índices de hipotiroidismo y obligan a descartarlo.
En el recién nacido:
- Peso mayor al de los recién nacidos normales.
- Retardo de la caída del cordón umbilical.
- Íctero fisiológico prolongado.
- No aparición de la epífisis distal del fémur o proximal de la tibia.
- Hipotonía muscular.
En los primeros 6 meses de la vida:
- Dificultad en la alimentación, que se manifiesta por pereza, falta de interés, somnolencia y crisis de sofocación durante la lactancia.
- Dificultad respiratoria, dada por episodios de apnea ruidosa y obstrucción nasal, a causa del aumento de tamaño de la lengua.
- Abdomen globuloso, en muchos, con hernia umbilical.
- Temperatura subnormal, piel fría y moteada (livedo reticular).
- Retraso psicomotor.
- Retraso de la edad ósea.
- Constipación.
- Llanto ronco.
En los mayores de 6 meses:
- Retraso del crecimiento.
- Proporciones infantiles del esqueleto (predominio del segmento superior sobre el inferior), no acordes con su edad.
- En la cabeza se encuentran:
- Fontanela anterior ampliamente abierta.
- Facies infantiles.
- Hipertelorismo.
- Nariz trilobulada con depresión de su raíz.
- Hendiduras palpebrales estrechas.
- Párpados tumefactos.
- Lengua gruesa y ancha que sobresale de la boca abierta.
- Retraso de la dentición y tendencia a las caries.
- Cuero cabelludo engrosado; cabellos ásperos, quebradizos.
- Mixedema, más ostensible en la cara, el dorso de las manos, los genitales externos y la región supraclavicular.
- Piel seca, fría y escamosa, transpiración escasa.
- Coloración amarilla de la piel por carotinemia.
- Pulso lento.
- Anemia.
- Hipotonía muscular.
Exámenes complementarios.
- Si el cuadro clínico es sugestivo de hipotiroidismo congénito el paciente debe ser evaluado inmediatamente y se determinará TSH y T4 en plasma. Solo un 5 % de los pacientes hipotiroideos puede ser sospechoso por sus manifestaciones clínicas. En el hipotiroidismo primario los niveles de TSH serán altos, y bajos los de T4.
- Si no son factibles los niveles plasmáticos de TSH y T4, se indica la determinación del yodo unido a la proteína (PBI) en la que se constarán valores bajos.
En la actualidad hay dos diagnósticos posibles en la etapa neonatal:
-
- Dosificar TSH al momento del nacimiento (mediante toma de muestra del cordón umbilical).
- Determinar T4, en este caso la extracción se realiza entre los días 3-5 de nacido.
- El retraso de la edad ósea constituye el más constante de los signos radiográficos esqueléticos; la disgenesia epifisaria puede ser característica, aunque no patognomónica. Existen signos radiográficos óseos que reflejan la inmadurez al nivel del cráneo y de la columna vertebral.
- Fosfatasa alcalina en sangre: Se constatan niveles bajos.
- Reflexoaquilograma: Se ha realizado en pacientes después del tercer año de vida, ya que necesita la cooperación del paciente; está prolongado en el hipotiroidismo sin tratamiento.
- Colesterol en suero: Se observan cifras altas. No es útil por debajo de los 2 años.
- En el hemograma puede encontrarse anemia.
- En el electrocardiograma del hipotiroidismo infantil, no predomina el bajo voltaje.
- Ecografía y gammagrafía tiroidea: Permiten el diagnóstico etiológico de las disembriogénesis tiroideas. La ecografía tiroidea en el recién nacido puede tener limitaciones técnicas, pero en manos expertas se ha llegado a visualizar el 79 % de las ectopias. La gammagrafía tiroidea se realiza en la actualidad con I123 o con Tc99, los que, al tener una vida media más corta que el I131 y al no incorporarse a la síntesis hormonal, producen una dosis menor de radioactividad.
Tratamiento.
Es de tipo sustitutivo y dura toda la vida, independientemente de la causa que origina el hipotiroidismo. Tiene como objetivo llevar al paciente rápidamente al estado eutiroideo, pues si no es instituido en tiempo y forma adecuados no podrá evitar el retraso mental y del crecimiento y desarrollo. Se utiliza la dosis máxima tolerada de hormonas tiroideas.
Recordar que la vida media de L-Tiroxina es de 7 días, deben ser ajustados los esquemas y administrar en una sola dosis al día.
El mejor índice de seguimiento es la evolución del paciente; la desaparición de los signos y síntomas de hipotiroidismo, así como el adecuado desarrollo físico, mental y óseo. Por tanto, el cuadro clínico y la curva de crecimiento y desarrollo son los datos más importantes en el seguimiento.
Cuando la administración de hormonas tiroideas es excesiva, puede aparecer insomnio, irritabilidad, taquicardia, cólicos abdominales, diarreas y aceleración de la edad ósea. En esos casos debe disminuirse la dosis.
Seguimiento.
Las consultas deben ser mensuales durante el primer año de tratamiento, trimestrales durante el segundo y semestrales a partir del tercer año de tratamiento.
En cada oportunidad se debe valorar:
- Ritmo de crecimiento y desarrollo.
- TSH y T4 en plasma: Los niveles elevados de TSH reflejan que la dosis de hormona tiroidea es insuficiente cuando se trata de pacientes con hipotiroidismo primario. Igualmente los valores evolutivos de T4 indican la dosis de L-Tiroxina Sódica.
- Colesterol.
- Reflexoaquilograma: Se ha utilizado en niños mayores de 3 años, los valores prolongados demuestran insuficiencia de tratamiento.
- Radiografía para determinar edad ósea; semestral los 2 primeros años, después, anual.
- Tiroglobulina: Se ha utilizado para detectar presencia de tejido tiroideo.
- Anticuerpos antitiroideos.
Es fundamental que los padres sean bien informados sobre la enfermedad, y sobre todo, de la importancia que tiene cumplir cabalmente el tratamiento durante toda la vida.
Es imprescindible conocer que no debe indicarse vitamina D ni calcio suplementario.
De existir dudas se realizarán pruebas terapéuticas, es decir, administrar hormonas tiroideas de la manera indicada durante los 3 primeros años de vida; después se debe suspender el tratamiento, observar clínicamente y valorar la función tiroidea. De presentarse algunos datos que afirmen el diagnóstico, es necesario reiniciar la terapéutica y mantenerla durante toda la vida; por el contrario, si al suspender el medicamento se comprueba de manera evidente que no se presentan manifestaciones de hipotiroidismo y el desarrollo y crecimiento no se alteran, se suspende definitivamente el tratamiento y se comunica a los familiares que el niño no presenta alteración alguna.
En el control evolutivo se debe vigilar el desarrollo neuropsíquico. Los test recomendados son el Brunet-Lezine hasta los 3 años, de 4 a 7 años el Terman-Merril o WIPSSI y el Wisc-R a partir de los 7 años.
En los casos de hipotiroidismo severo por diagnóstico tardío es preferible hospitalizar al niño durante los primeros 10 días, para comenzar con el tratamiento y evitar una posible descompensación cardiaca.
Detección neonatal de fenilcetonuria.
La fenilcetonuria es uno de los errores congénitos del metabolismo de los aminoácidos, debido al déficit de la enzima fenilalanina hidroxilasa hepática. Esta enzima cataliza la reacción de hidroxilación de la fenilalanina a tirosina. El déficit de la enzima provoca niveles de fenilalanina en suero treinta veces superiores a lo normal y existe un incremento en la excreción urinaria del aminoácido. El nivel de fenilalanina en el líquido cefalorraquídeo está también considerablemente elevado.
Las concentraciones elevadas de fenilalanina dan lugar a varios trastornos bioquímicos secundarios. En los paciente no tratados estos trastornos provocan daño cerebral postnatal con el consiguiente retraso mental. Un grupo de reacciones involucra cambios en la cadena lateral de la fenilalanina y grandes cantidades de ácido fenilpirúvico y ácido fenilacético. El ácido fenilacético se conjuga subsecuentemente con la glutamina para formar fenilacetilglutamina. Estos metabolitos son excretados por la orina.
Esta enfermedad sigue un patrón de herencia AR y a pesar de no tener una frecuencia elevada en nuestro país (1 en 40 000 aproximadamente) en comparación con otras enfermedades de origen genético, la posibilidad de tratamiento efectivo con la consiguiente prevención del retraso mental justifica su pesquisa masiva.
Cuando existe descompensación de la enfermedad, comienza a aparecer coloración más clara en la punta del cabello, manifestación clínica fundamental para el seguimiento de estos pacientes. (Cuadro 3)
El tratamiento de esta condición ha estado dirigido a restringir el contenido dietético de fenilalanina. Para la realización del pesquisaje se toma una muestra de sangre de todos los neonatos al quinto día de nacidos por punción del talón colectada en papel de filtro y con los requisitos establecidos para la toma de muestra (ver más adelante).

La fenilalanina es un aminoácido esencial que se obtiene a través de la dieta, por lo tanto hay que esperar un margen de tiempo durante el cual el neonato puede adquirirlo, fundamentalmente por la lactancia materna, y de esta forma la existencia del déficit enzimático explicará el aumento de la fenilalanina en sangre. En aquellos casos a los cuales no se les realiza la prueba antes del día 15 de vida puede realizarse con posterioridad, pues es preferible el diagnóstico tardío para evitar un retraso mental más profundo por ausencia de diagnóstico. No obstante, la eficiencia del tratamiento depende en gran medida de su precocidad.
Cifras de fenilalanina en suero entre 4 y 10 mg indican seguimiento de los pacientes que generalmente evolucionan hacia la normalidad y son asintomáticos. Se considera que existe hiperfenilalaninemia cuando las cifras se encuentra entre los 10 y 20 mg/dL mientras que el diagnóstico positivo de la enfermedad se realiza a todos aquellos pacientes con niveles plasmáticos de fenilalanina superiores a 20 mg/dL en el periodo neonatal, asociado a niveles normales o disminuidos de fenilalanina ante hablactaciones normales.
Los neonatos en los que se diagnostican cifras superiores a 10 mg/dL son seguidos por consultas multidisciplinarias suministrándosele la dieta adecuada que evita la aparición de síntomas.
Es importante recordar que esta enfermedad autosómica recesiva tiene un riesgo de recurrencia de 1:4, lo que indica que por la alta probabilidad de repetición de la entidad, las parejas que han tenido un hijo afectado son tributarias de atención por los servicios de genética ante nuevos embarazos.
Detección neonatal de hiperplasia adrenal congénita.
La hiperplasia adrenal congénita (HAC) es un síndrome adrenogenital producido por un bloqueo en la síntesis del cortisol.
Resulta de tres alteraciones:
- Falta de cortisol: Las glándulas adrenales de las personas con HAC tienen insuficientes cantidades de una de las enzimas que se necesitan para producir cortisol. El cuerpo que tiene deficiencia de cortisol no puede movilizar suficiente azúcar. El nivel deficiente de azúcar puede causar somnolencia y coma.
- Pérdida de sal: En algunos niños con HAC se pierden grandes cantidades de sal en su orina. Un gran desequilibrio de sal podría causar vómitos seguidos de deshidratación, debilidad muscular extrema y debilidad en el músculo del corazón, la pérdida de sal ocurre en un 75 % (3 de cada 4) de los niños con HCA.
- Exceso de andrógeno: La adrenal estimulada no puede producir cortisol, pero continúa produciendo demasiadas hormonas masculinas.
La forma más común de HCA es la deficiencia de la enzima 21 hidroxilasa, responsable del 90-95 % de los casos, esta deficiencia se traduce en un aumento de los niveles de 17 OHP, principal sustrato de la 21 hidroxilasa.
Clasificación de la HAC:
- Forma clásica.
- Forma no clásica o de aparición tardía.
El 80 % de los casos de HAC corresponden con la forma clásica de la enfermedad. Su presentación ocurre en el periodo neonatal, y resulta en un bloqueo completo o casi completo de actividades enzimáticas. Dentro de esta forma se encuentran dos subclases de pacientes:
- Perdedores de sal.
- Virilizantes simples.
La forma clásica perdedora de sal es la más severa, ocupa el 75 % de los casos de la forma clásica y deriva en deficiencia de cortisol y aldosterona, exceso en la producción de andrógenos adrenales desde la vida fetal, virilización de genitales externos en los recién nacidos de sexo femenino y alteraciones por deficiencia de aldosterona.
Manifestaciones clínicas
En las niñas recién nacidas con este trastorno, el clítoris esta agrandado y tiene la abertura de la uretra en la base (genitales ambiguos), las estructuras internas del tracto reproductivo (ovarios, útero, trompas de Falopio) son normales. Al avanzar en edad, se produce la masculinización de algunos de sus rasgos, tales como engrosamiento de la voz, aparición de vello facial y ausencia de menstruación en la pubertad.
En los niños recién nacidos no se presenta ninguna anomalía aparente, pero mucho antes de que ocurra normalmente la pubertad el niño se vuelve cada vez más musculoso, se agranda el pene, aparece el vello púbico y la voz se vuelve más grave. Los varones afectados parecen iniciar una pubertad muy prematura entre los 2 y 3 años de edad. En la pubertad, los testículos son pequeños. Algunas formas de HAC son más severas y ocasionan crisis suprarrenales en el recién nacido debido a la pérdida de sal. En esta forma de HAC donde se pierde sal, los recién nacidos desarrollan síntomas poco después de nacer, por lo general, vómitos, deshidratación, cambios electrolíticos y arritmias cardiacas. De no tratarse esta condición, el bebé puede morir entre 1 a 6 semanas después de haber nacido.
Los síntomas y signos en los recién nacidos, incluyen inapetencia, letargia, diarreas, deshidratación, hipotensión y pérdida de peso; en los no tratados, son inminentes el colapso circulatorio y la muerte. Como el control de líquidos y electrolitos en el feto es mantenido por la placenta, esta «crisis perdedora de sal», se desarrolla sólo después del nacimiento, usualmente desde la primera hasta la cuarta semana de vida, la acumulación de altas concentraciones de 17 OHP con leves propiedades natriuríticas, puede exacerbar la pérdida de sal.
La inhabilidad para retener sodio y excretar potasio de los túbulos renales por deficiencia de aldosterona resulta en hiponatremia, hiperpotasiemia, acidosis metabólica y en algunos casos hipoglucemia por deficiencia de cortisol.
Esta enfermedad se hereda de forma autosómica recesiva.
Diagnóstico prenatal
En general el diagnóstico prenatal se realiza en familias donde han existido casos de niños con HAC, se estudian los embarazos posteriores buscando la posible existencia de la enfermedad a través de la amniocentesis o por uso de muestras de vellosidades coriónicas en el primer y segundo trimestre respectivamente; si se confirma la enfermedad puede iniciarse el tratamiento antes del nacimiento.
Se ha propuesto que el líquido amniótico extraído por amniocentesis transabdominal para el diagnóstico debe realizarse a las 16 semanas, ya que los niveles de 17 OHP se encuentran ligeramente disminuidos entre las 11 y 15 semanas con un máximo en las 14 semanas.
Con el análisis del ADN en células fetales, el diagnóstico prenatal de la deficiencia de la 21 Hidroxilasa, o el estado de portador de los genes relacionados con dicho trastorno, puede determinarse con un 95 % de exactitud.
Diagnóstico neonatal
Técnicas de inmunoensayo han permitido el pesquisaje neonatal de la HAC empleando muestras de sangre fresca sobre papel de filtro. En la actualidad la determinación de los niveles elevados de 17 OHP, en muestra de sangre seca sobre el papel de filtro es mundialmente usada para el diagnóstico de la HAC. El médico o enfermera de la familia indicará el estudio a todos los neonatos el quinto día de nacidos. En este sentido es muy importante que se analice la posibilidad de la HAC perdedora de sal, cuyas manifestaciones aparecen en las primeras semanas de la vida. Este estudio se realizará en el mismo papel de filtro que se utiliza para la pesquisa de fenilcetonuria.
El diagnóstico puede ser confirmado mediante una elevación hormonal donde se incluye la determinación de los niveles de 17 OHP en muestra de suero, el perfil de esteroides en orina y los niveles de pregnantiol en orina colectada durante 24 horas. Pueden emplearse también pruebas de estimulación hormonal donde se determinen los niveles de 17 OHP antes y después de la estimulación con hormona adrenocorticotrópica (ACTH).
El propósito del tratamiento de la HAC es la sustitución del cortisol y la aldosterona con el objetivo de normalizar los niveles de la síntesis de ACTH y de esa manera disminuir la producción de andrógenos.
El tratamiento para los pacientes con HAC en el período neonatal consiste en la administración de glucocorticoides y de mineralocorticoides.
Detección neonatal de déficit de biotinidasa.
La deficiencia de biotina congénita se debe a una deficiencia de la biotinidasa, enzima que participa en el ciclo de la biotina.
Los síntomas clínicos que acompañan la deficiencia de biotinidasa son convulsiones aisladas o asociadas a otros síntomas neurológicos, hipotonía, ataxia, y pérdidas de visión y audición. Los síntomas dermatológicos son erupción cutánea y alopecia. Los pacientes también suelen presentar cetoacidosis e hiperamonemia. La administración de biotina exógena corrige estos síntomas.
La deficiencia de biotinidasa es una de las enfermedades metabólicas congénitas que es prevenible y tratable. Además, es una situación absolutamente excepcional: se produce un caso por cada 150 000 nacimientos.
En el metabolismo intervienen cuatro carboxilasas. Cuando existe una deficiencia en estas carboxilasas se altera el metabolismo y es deficiente la síntesis de ácidos grasos y la glucogénesis; así, se acumulan una serie de productos que resultan tóxicos y que actúan, fundamentalmente, sobre el sistema nervioso central, dando lugar a un cuadro extraordinariamente complejo.
Si un recién nacido tiene un déficit de biotinidasa, durante los primeros tiempos de su vida la madre le aporta biotina y no aparecen síntomas clínicos, pero según el niño va creciendo, aparecen síntomas evidentes y alarmantes.
La pesquisa neonatal se realiza a los 5 días de nacido, la muestra de sangre del talón se colecciona en el mismo papel de filtro que se usa para el estudio fenilcetonuria, galactosemia e hiperplasia adrenal congénita. Por ello es necesario que la toma de muestra de sangre del talón se haga con la calidad requerida.
El tratamiento específico está basado en la administración de biotina (20 mg diarios).
Detección neonatal de galactosemia.
La galactosemia es una enfermedad congénita del metabolismo de los carbohidratos, se hereda por un gen recesivo autosómico. Es la incapacidad del organismo para utilizar (metabolizar) el azúcar en simple galactosa (causante de la acumulación de galactosa 1 fosfato), que alcanza altos niveles en el organismo y llega a causar afectación en varios órganos.
Su frecuencia es aproximadamente de 1 por cada 60 000 nacimientos.
Existen tres formas de la enfermedad:
- Deficiencia de galactosa-1-fosfatouridil transferasa (galactosemia clásica, la forma más común y más grave).
- Deficiencia de galactosa-quinasa.
- Deficiencia de galactosa-6-fosfato epimerasa.
Si a un bebé con galactosemia se le da leche, los derivados de la galactosa se acumulan en su sistema, causando daño al hígado, al cerebro, a los riñones y a los ojos. Los individuos con galactosemia no pueden tolerar ninguna forma de leche (ni humana ni animal) y deben vigilar cuidadosamente la ingesta de otros alimentos que contengan galactosa. La exposición a los productos lácteos puede ocasionar daño hepático, retardo mental, formación de cataratas e insuficiencia renal.
Después de tomar leche durante algunos días, un neonato con galactosemia desarrollará intolerancia a la alimentación, ictericia, letargia, irritabilidad y convulsiones. Así mismo, se presentará agrandamiento del hígado y el azúcar puede estar bajo.
Diagnóstico neonatal
Esta es otra de las enfermedades que se estudian en el mismo papel de filtro de la fenilcetonuria, por lo que la toma de muestra se realiza a los cinco días de nacido, por el médico o la enfermera de la familia. El estudio de la muestra se realiza por técnicas del sistema ultramicroanalítico (SUMA). Se realiza seguimiento a partir de 10 mg % en muestra de talón.
Se considera positivo el estudio de esta enfermedad cuando los valores de galactosa en sangre son mayores de 4,3 mg %.
Cuando se obtienen estos valores se cuantifican los valores de galactosa en orina, que serán superiores a 8 mg %.
Tratamiento
Una vez que se diagnostica la enfermedad, se realiza un tratamiento que consiste en la abstinencia estricta del consumo de todos los tipos de leche y los productos lácteos. El niño puede ser alimentado con fórmulas basadas en soja, carne o Nutramigen (una fórmula procesada a base de proteína hidrolizada). Los padres deben cuidar de su hijo y enseñarle a evitar no sólo la leche y derivados lácteos, sino también los productos que contengan leche en polvo, por lo cual es esencial mantenerse informado y leer las etiquetas de los productos.
Manejo nutricional
- Suministrar el aporte energético proteico adecuado.
- Suplementar con vitaminas y minerales.
- Educación nutricional a padres y familiares.
- Control dietético estricto.
El tratamiento dietético puede administrarse por vía oral o parenteral (pacientes en estado crítico):
Oral: De 0 a 3 meses: Fórmula basal de carne y jugos de frutas naturales. Leches de fórmula de aislado de proteína de soya; de 3 a 12 meses: Puré de viandas, leguminosas, vegetales, cereales y frutas frescas; a partir de 12 meses: Dieta libre exenta de productos lácteos y sus derivados.
Tratamiento parenteral: Expectativas (pronóstico): El niño puede vivir una vida normal, si se hace un diagnóstico temprano y se evitan estrictamente los productos lácteos.
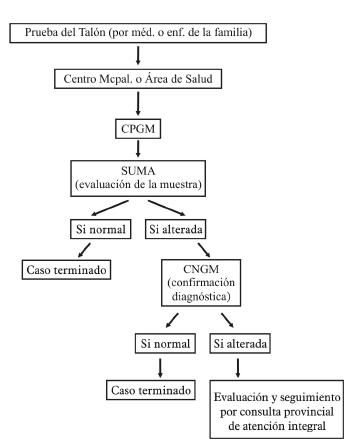
La toma de la muestra del talón se debe realizar por el médico o enfermera de la familia y se traslada al Centro Municipal o Área de Salud, luego al Centro Provincial de Genética Médica y seguidamente al laboratorio del SUMA.
Ventajas que ofrece la colecta de sangre en papel de filtro
- Simplifica el traslado de las muestras desde lugares distantes hasta un laboratorio central sin necesidad de refrigeración ni cuidados especiales.
- Implica un menor riesgo biológico.
- Permite fácil almacenamiento y conservación sin que se afecte la estabilidad.
- No se requiere de punción venosa y la cantidad de sangre necesaria es pequeña.
Técnicas para la colección, secado y traslado de la muestra de sangre
El niño debe ser cargado de forma que sus piernas estén más bajas que el corazón para que se incremente la presión venosa.
Se limpia el sitio de punción con algodón estéril y alcohol al 70 % y luego secado de la zona.
Con una lanceta estéril se punciona la porción media o lateral de la superficie plantar del talón.
Se elimina con una gasa estéril la primera gota (si quedara algún residuo de alcohol se puede diluir la muestra y afectar los resultados).
La gota de sangre debe ser lo más grande posible para que garantice el llenado del área y embeber el papel de filtro de forma tal que la mancha de sangre se observe por ambas caras del papel.
No se debe exprimir el sitio de punción porque puede causar hemólisis y además mezcla de tejidos con la muestra).
El papel se debe tocar suavemente con la gota y nunca presionarlo contra el talón buscando una mancha mayor.
La sangre debe ser aplicada por una sola cara; habrá que asegurarse de que traspase el papel.
No deben aplicarse sucesivas gotas sobre el círculo, pues se formaría más de una capa de sangre y/o concentraciones no uniformes de la sangre.
Después que la sangre ha sido colectada del talón del recién nacido, el pie debe ser elevado por encima del cuerpo y presionar con una gasa estéril o algodón contra el sitio de punción hasta que deje de sangrar.
Las muestras deben secarse al menos 3 horas a temperatura ambiente colocadas en posición horizontal sin tocar superficie alguna.
No se deben mover ni calentar durante el secado.
No exponer al sol porque la temperatura alta desnaturaliza proteínas y las fija al papel.
Después de estar bien secas, las muestras deben ser trasladadas en sobres de papel con correcta identificación, no en nylon herméticamente cerradas. Se aplicarán las normas de bioseguridad establecidas para la transportación de material biológico.
Deben ser enviadas al Centro Provincial de Genética dentro de las 24 horas después de su recogida para control de la calidad y traslado al laboratorio SUMA para su procesamiento.
Causas de error en los resultados del pesquisaje de la deficiencia de biotinidasa
- Pérdida de la actividad de la enzima en muestras de sangre seca sobre papel de filtro, almacenadas a temperatura ambiente.
- Interferencias que se producen en la señal de ensayos colorimétricos debidas al uso de determinados fármacos.
Causas de error en los resultados del pesquisaje de galactosemia
- Muestras tomadas después de una transfusión sanguínea.
- Pacientes alimentados con fórmulas basadas en sojas o sin lactosa.
- Muestras tomadas antes de las 48 horas de nacido.
- Inadecuada ingesta de leche.
- Hiperbilirrubinemia severa.
El médico y la enfermera de familia en el área de salud deben ser cuidadosos en el cumplimiento del test neonatal, tanto en la realización de este a todos sus recién nacidos, como en que se realice con la calidad requerida, siguiendo las normas técnicas para ello.
El Área de Salud debe velar porque las muestras en papel de filtro lleguen a su destino sin dificultades.
CONCLUSIONES
El diagnóstico y prevención de las enfermedades genéticas en la atención primaria de salud es un tema complejo por el compromiso que implica su correcto desarrollo y sistematización. La actualización y la coherente preparación de los profesionales de este nivel de asistencia médica en estos temas, es fundamental para el desarrollo óptimo de las acciones de este programa. Por ello la utilidad de este folleto como material bibliográfico en la práctica del asesoramiento genético.
Summary
Essentials of diagnosis and prevention of genetic diseases in the primary health care.
Programs aimed at preventing genetic diseases reduce their fatal consequences among the population. Therefore, it is important to correctly prepare the medical staff working in the Maternal-Infant Program. Subjects related with these issues are thought since the pre-graduate education, in the second year of Medicine studies and in the postgraduate courses as part of the General Comprehensive Medicine residency within the subject Medical Genetics. The importance of this topic supported the creation of this pamphlet, based on a bibliographical review of the subject related with the implementation of the diagnosis and prevention program for genetic disease within the practice of genetic advising. The objective of this pamphlet is to provide the basic theoretical elements to facilitate the implementation of health actions in the community, which can be further review using the conventional bibliography.
Key words: Genetic diseases, inborn;genetics, medical;genetic services;diagnosis;disease prevention;
Referencias bibliográficas
1. Álvarez Fumero R. Enfermedades heredometabólicas y embarazo. Rev Cubana Obstetricia Ginecología [Serie en Internet]. 2003[ cited 3 Ene 2005]; 29(2): Avaidable from: http://www.bvs.sid.cu/revis tas/gin/vol29_2_03/gin01203.htm.
2. Campisol J. Errores congénitos del metabolismo intermediario con repercusión neurológica. Aminoacidopatías. Acidurias orgánicas. En: Glaxo Welcome: Neurología Pediátrica. Madrid: Ergon; 2000. p. 95-113.
3. Carvalho HB, Brizot ML, Lopez LM, Chiba CH, Meyadahira H, Zugaib M. Detection of fetal structural abnormalities at the 11-14 week Ultrasound scan. Prenat Diagn. 2002; 22:1-4.
4. Campistol J, Lambruschini N, Vilaseca MA, Cambra FJ, Fuste E, Gómez L. Hiperfenilalaninemias. En: Sanjurjo P, Baldellou A (ed.). Diagnóstico y tratamiento de las enfermedades heredometabólicas hereditarias. Madrid: Ergon; 2001. p. 195-206.
5. Del Monte E, Viñas Portilla C, González Garcías N, Lantigua Cruz A. Reflexiones sobre la atención a personas con defectos genéticos en el nivel primario de salud. Rev Cubana Med Gen Integr. 2009; 16(2):194-7.
6. Documento Normativo de la Comisión Nacional de Atención a Fenilcetonúricos. La Habana: INA; 2000. p. 4-7.
7. Ezquieta B, Cueva López E, Varela Junquera JM, Jariego Fente C. Aportaciones del análisis molecular en la hiperplsia suprarrenal congénita. Acta Pediatr Esp. 2001; 59:479-96.
8. Fernández J, Sadrubay JM, Van den Berhe G. Inborn Metabolic Disease: diagnosis and treatment. 3ra ed. Berlín: Springer Verlag; 2000.
9. González Jiménez G, Gómez Baute R, González Iglesias Y. Evaluación de la eficacia diagnóstica por ultrasonografía en malformaciones congénitas mayores. Rev Cubana Obstet Ginecol [Serie en Internet]. 2002[ cited 8 May 2005]; 8(3): [aprox. 9p]. Avaidable from: www.bvs.sld.cu/revistas/gin/vol28_3_02/gin01302.htm.
10. Gutierrez García E, Barrios García B, Carrillo Estrada U. Galactosemia. Diagnóstico precoz mediante estudio enzimático. Rev Cubana Pediatría [Serie en Internet]. 2002[ cited 7 May 2005]; 75(3): Avaidable from: http://scielo_sld.cu/cgi-bin/wxis.exe/iah/IsisScrip=iah/iah.xis&base=artic.
11. Gutierrez García E, Barrios García B, Carrillo Estrada U. Galactosemia: diagnóstico de 5 casos. Rev Cubana Pediatría [Serie en Internet]. 2001[ cited 7 May 2005]; 73(2): [aprox. 8p]. Avaidable from: http://scielo_sld.cu/cgi-bin/wxis.exe/iah/IsisScrip=iah/iah.xis&base=artic.
12. Labarta JI, Belloa E, Ruiz-Echarrib M, Ruedab C, Martulc P, Mayayod E, et al. Estado en la edad adulta y propuesta de optimización terapéutica de la hiperplasia suprarrenal congénita. Anales de Pediatría [Serie en Internet]. 2003[ cited 16 May 2005]; 58(12): [aprox. 15p]. Avaidable from: www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PutMed&list_inds=21837830&dopt=Abstract.
13. Mueller RF, Young ID. Screening de poblaciones y genética de comunidades. En: Emery´s. Genética Médica. 10ma ed. Madrid: Marban; 2001. p. 311-18.
14. Martín Ruiz MR. Programa de prevención de anemia falciforme (III). La electroforesis de hemoglobina: indicación e interpretación. Rev Cubana Medicina General Integral [Serie en Internet]. 1996[ cited 29 Abr 2005]; 12(2): [aprox. 12p]. Avaidable from: http://www.bvs.sld.cu/revistas/mgi/vol12_2_96/mgi06296.
15. Mueller RF, Young I. Hemoglobinas y hemoglobinopatías. En: Emery´s. Genética Médica. 10ma. ed. Madrid: Marban; 2001. p. 137-48.
16. Mueller RF, Young I. Diagnóstico prenatal de desórdenes genéticos. En: Emery´s. Genética Médica. 10ma. ed. Madrid: Marban; 2001. p. 301-9.
17. Mueller RF, Young I. Genética del desarrollo. En su: Emery´s. Genética Médica. 10ma. ed. Madrid: Marban; 2001. p. 81-95.
18. Mueller RF, Young I. Alteraciones cromosómicas. En: Emery´s. Genética Médica. 10ma. ed. Madrid: Marban; 2001. p. 246-62.
19. Mariño M, Zarzalejo Z. Tratamiento nutricional de un niño con fenilcetonuria de diagnóstico neonatal: Estudio del caso. An Venez Nutr. 2000; 13(1):202-9.
20. Merke D, Bornstein SR, Avila NA, Chrousos GP. Future directions in the study and management of congenial adrenal hyperplasia due to 21-hydroxylase deficiency. Ann Intern Med. 2002; 136:320-34.
21. Oliver A, Ezquieta B, Gussinyé M. Hiperplasia suprarrenal congénita. En: Argente J, Carrascosa A, Gracia R, Rodríguez Hierro F. (eds). Tratado de Endocrinología Pediátrica y de la Adolescencia. 2da ed. Barcelona: Doyma; 2000. p. 995-1042.
22. Puertas Bordallo D, Martín Reyes C, Ruiz-Falcó Rojas ML, Duat Rodrígues A, Valls Ferrán, MI. Neuropatía óptica por déficit de biotinidasa. Rev Cubana de Pediatría [Serie en Internet]. 2004[ cited 3 Jun 2005]; 8: [aprox. 7p]. Avaidable from: http://www.oftalmo.com/seo/2004/08ago04/07.htm.
23. Sellers López F. Diagnóstico precoz de los defectos del tubo neural. Rev Inst Bernabeu, Alicante. 1999; 97:132-133.
24. Sheard NF. Importance of diet in maternal phenilketonuria. Nutr Rev. 2000; 58(8):236-9.
25. Umtest PKU. Prueba fluorescente para la cuantificación de Phe en sangre seca sobre papel de filtro. La Habana: Centro de Inmunoensayo; 2004.
26. Speiser PW. Congenital adrenal hyperplasia owing to 21-hydroxylase deficiency. Endocrinol Metab Clin North Am. 2001; 30:31-60.
27. White PC, Speiser PW. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocr Rev. 2000; 21:245-91.
28. White PC, Speiser PW. Long term consequences of childhood-onset congenital adrenal hyperplasia. Best Pract Res Clin Endocrinol Metab. 2002; 16:273-88.
29. Umelisa. 17 OH Progesterona neonatal para la determinación cuantitativa de 17 OH Progesterona en sangre fresca sobre papel de filtro. La Habana: Centro de Inmunoensayo; 2008.
30. Umtest biotinidasa. Prueba colorimétrica para la detección de biotinidasa en sangre seca sobre papel de filtro. La Habana: Centro de Inmunoensayo; 2007.
31. Umtest galactosemia. Prueba enzimática y fluorescente para la cuantificación de galactosa total en sangre seca sobre papel de filtro. . La Habana: Centro de Inmunoensayo; 2007.
Recibido: 26 de noviembre de 2008. Aprobado: 20 de mayo de 2009.
El ABC para el diagnóstico y prevención de las enfermedades genéticas en la atención primaria de salud. Facultad de Ciencias Medicas Cienfuegos. Calle 51A y Avenida 5 de Septiembre. Cienfuegos, Cuba. CP 55100. Email: mikhail@infomed.sld.cu
1Especialista de I Grado en Medicina General Integral. M Sc. en Asesoramiento Genético. Policlínico Área III. Cienfuegos
2Especialista de I Grado en Genética Clínica. Profesor Auxiliar. Hospital Pediátrico ?Paquito González Cueto?. Centro Provincial de Genética.
3Especialista de I Grado en Laboratorio Clínico. Profesor Asistente. Hospital Pediátrico ?Paquito González Cueto?
4Especialista de I Grado en Genética Clínica. Hospital Pediátrico ?Paquito González Cueto?
5Especialista de I Grado en Pediatría. Hospital Pediátrico ?Paquito González Cueto?
6Especialista de I Grado en Medicina General Integral. M Sc. en Asesoramiento Genético. Policlínico de Lajas. Cienfuegos.
