










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkMediSur
versión On-line ISSN 1727-897X
MediSur v.8 n.4 Cienfuegos jul.-ago. 2010
Presentaciones de casos
Revista Científica de las Ciencias Médicas en Cienfuegos
Síndrome de Apert. Reporte de un caso
Ninecta Pérez Breña,1 Francisco Amed Abad Aguiar,2 Dunia Reyes Hernández,3 Yamil González Martínez,4
Resumen
Se presenta el caso de una paciente de sexo femenino, de color de piel blanca, de 7 años de edad, evaluada en el Servicio de Atención Primaria de la misión médica Barrio Adentro del estado Nueva Esparta, en la República de Venezuela. Después de la evaluación clínica y radiológica se diagnostica un síndrome genético conocido como Síndrome de Apert.
Palabras clave: Acrocefalosindactilia;radiología;diagnóstico;
Summary
Apert Syndrome. Case Report
The case of a white female aged 7 is evaluated in the Primary Care Service of the Barrio Adentro medical mission in Nueva Esparta state, Republic of Venezuela. After a clinical and radiological evaluation she is diagnosed with a genetic syndrome known as Apert Syndrome.
Key words: Acrocephalosyndactylia;radiology;diagnosis;
INTRODUCCIÓN
El síndrome de Apert, conocido como acrocéfalo-sindactilia, es una rara enfermedad hereditaria con patrón autosómico dominante, cuyo defecto se produce por una mutación espontánea de origen paterno, que afecta al receptor 2 del factor de crecimiento de los fibroblastos (FGFR2). Se calcula una prevalencia de 15 por 1 millón de nacidos vivos, aunque es mayor en los países asiáticos donde puede llegar a 22,3. La relación entre la edad del padre y la aparición de la mutación es directamente proporcional. (1-4)
El defecto genético produce un cierre precoz de las suturas craneales, con la subsecuente aparición de craneosinostosis, lo que ocasiona un crecimiento asimétrico de la cabeza, además de una sindactilia simétrica que afecta frecuentemente los cuatro miembros, se acompaña de dismorfia facial y alteraciones neurales. Es frecuente que aparezca el retardo mental. (1, 4)
Los pacientes y sus familiares deben ser evaluados por equipos multidisciplinarios. Se hace necesaria la programación de un grupo de intervenciones quirúrgicas que comienzan en la infancia, etapa en la que se realiza la craneotomía descompresiva. Existen otras intervenciones con las que se puede disminuir la obstrucción de las vías aéreas, las operaciones estéticas y funcionales como las faciales, las odontológicas y la de los miembros (separación de dedos de manos) que pueden realizarse en la infancia tardía y en la adolescencia. La familia debe ser apoyada psicológicamente y así mismo debe ser entrenada para el manejo adecuado del caso para de esta forma lograr la incorporación de estas personas a la sociedad y para evitar la marginación a que puede ser expuesto el paciente. (1, 4-9)
La realización de estudios ultrasonográficos ayudan al diagnóstico prenatal, con ellos se detectan anomalías que son sugestivas del Síndrome de Apert, lo que puede ser confirmado con la realización de una amniocentesis, previo consentimiento de los padres, los cuales deben estar impuestos del pronóstico del caso. (10)
PRESENTACIÓN DEL CASO
Se presenta el caso de una paciente de sexo femenino, de color de piel blanca, de 7 años de edad, que nació de un embarazo normal, a término (parto fisiológico) que pesó al nacer 2 488 g, no presentó íctero, ni cianosis, ni tampoco otras complicaciones peri o postnatales.
Recibió lactancia materna hasta los cinco meses, luego lactancia mixta. Presentó retardo en el desarrollo psicomotor, la sonrisa social la presentó a los 6 meses, no gateó, el brote de la primera dentición se produjo a los once meses y pronunció las primeras palabras a los 14 meses. No se recogieron antecedentes de casos similares en la familia.
La paciente fue llevada a la consulta integral de atención primaria de la misión médica Barrio Adentro del estado Nueva Esparta, Venezuela. La madre refirió que durante el embarazo no sufrió alteraciones psicológicas, ni recibió tratamiento farmacológico o sufrió trauma físico alguno.
Al examen físico se observó cráneo alargado en forma de barquilla, alteraciones oculares con hipertelorismo, exoftalmos y órbitas aplanadas, cara con ojos saltones, aplanamiento de los huesos frontal y occipital, el tercio medio de la cara estaba aplanado e hipodesarrollado, nariz picuda, oblicuidad antimongoloide de las hendiduras palpebrales. Se observó además al examen bucal apelotonamiento de los dientes. (Figura 1).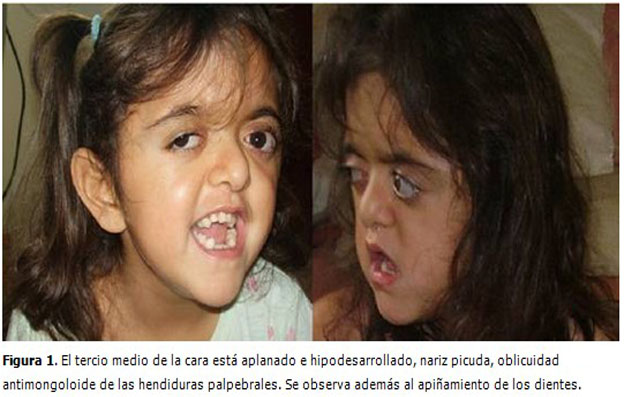
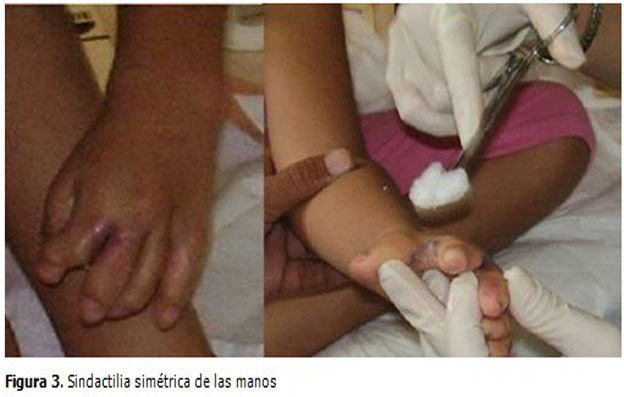
Las manos y los pies estaban deformados simétricamente, presentaba sindactilia en las manos con fusión de los dedos índice, medio y anular mientras que en los pies es con unión de todos los dedos. (Figuras 2 y 3).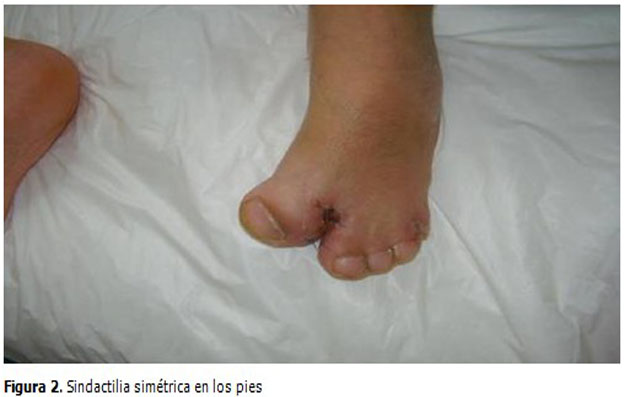
Al realizarse la evaluación multidisciplinaria se determinó la presencia de afectación severa del lenguaje y moderada de la inteligencia, no se encontraron anormalidades cardiovasculares ni en otros sistemas.
Exámenes radiográficos:
Los rayos X realizados en las manos mostraron: sindactilia ósea y de los tejidos blandos de los dedos II-III-IV en ambas manos.
La conjunción de todos los signos clínicos hallados permitió llegar a la conclusión que se estaba en presencia de una paciente con Síndrome de Apert.
Como parte de la evaluación multidisciplinaria la paciente fue referida al Servicio de Odontología, se realizó también interconsulta con el Servicio de Ortopedia Infantil para valorar una posible cirugía de las alteraciones de las manos y de los pies, como parte del mejoramiento de la calidad de vida de la paciente. Se brindó a la madre apoyo psicológico y orientación adecuada para lograr la incorporación social de esta paciente.
Referencias bibliográficas
1. DeGiovanni C, Jong C, Woollons A. What syndrome is this? Apert syndrome. Pediatr Dermatol. 2007; 24(2):186-8.
2. Fanganiello R, Sertié A, Reis E, Yeh E, Oliveira N, Bueno D, et al. Apert p.Ser252Trp mutation in FGFR2 alters osteogenic potential and gene expression of cranial periosteal cells. Mol Med. 2007; 13(7-8):422-42.
3. Carneiro G, Farias J, Santos P, Lamberti P. Apert syndrome: review and report a case. Rev Bras Otorrinolaringol. 2008; 74(4):583-87.
4. Yoon S, Qin J, Glaser R, Wang JE, Wexler N, Sokol R, et al. The ups and downs of mutation frequencies during aging can account for the apert syndrome paternal age effect. PLoS Genet. 2009; 5(7):998-1006.
5. Carro E, Fernández S. Síndrome de Apert. Presentación de un caso. Rev Cubana Pediatr. 2005; 77(3-4):
6. Moreno A, Chovil K, Forero J, Oyuela M. Sindrome de Apert. Rev colomb radiol. 2001; 12(4):1027-9.
7. Marucci D, Dunaway D, Jones B, Hayward R. Raised intracranial pressure in Apert syndrome. Plast Reconstr Surg. 2008; 122(4):1162-8.
8. Hohoff A, Joos U, Meyer U, Ehmer U, Stamm T. The spectrum of Apert syndrome: phenotype, particularities in orthodontic treatment, and characteristics of orthognathic surgery. Head Face Med. 2007; 8(3):10.
9. Salazard B, Casanova D. The Apert´s syndrome hand: therapeutic management. Chir Main. 2008; 27(Suppl 1):115-20.
10. Athanasiadis A, Zafrakas M, Polychronou P, Florentin-Arar L, Papasozomenou P, Norbury G, et al. Apert syndrome: the current role of prenatal ultrasound and genetic analysis in diagnosis and counselling. Fetal Diagn Ther. 2008; 24(4):495-8.
Recibido: 24 de diciembre de 2009. Aprobado: 04 de agosto de 2010.
Síndrome de Apert. Reporte de un caso. Facultad de Ciencias Medicas Cienfuegos. Calle 51A y Avenida 5 de Septiembre. Cienfuegos, Cuba. CP 55100. Email: ninec2005@yahoo.com
1Especialista de I Grado en Medicina General Integral.Sociedad Cubana de Medicina Familiar.
2Especialista de I Grado en Medicina General Integral. Profesor Instructor.Sociedad Cubana de Medicina Familiar.
3Especialista de II Grado en Histología. Máster en Ciencias. Profesor Auxiliar.Sociedad Cubana de Medicina Familiar.
4Especialista de I Grado en Medicina General Integral. Profesor Asistente.Sociedad Cubana de Medicina Familiar.
