










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkMediSur
versión On-line ISSN 1727-897X
Medisur vol.9 no.4 Cienfuegos jul.-ago. 2011
PRESENTACIÓN DE CASO
Queratodermia palmoplantar de Unna Thost. Presentación de un caso
Unna Thost Keratoderma palmoplantar . A Case Report
Graciela Caridad Cabrera AceaI , María Teresa Rodríguez GandullaII , Niurka Ramos VieraIII
I Policlínico Laboral Universitario. Área 7, Cienfuegos, Cienfuegos, Cuba
II Policlínico Universitario, Abreus, Cienfuegos, Cuba
III Policlínico Universitario. Área 1, Cienfuegos, Cienfuegos, Cuba
RESUMEN
Se presenta el caso de un paciente de color de piel negra, sexo masculino y de 37 años de edad que acudió a consulta del Servicio de Dermatología de su área de salud por presentar lesiones cutáneas en las palmas de las manos y en las plantas de los pies. Refirió que las lesiones se agudizaron con el tiempo. Estas se convirtieron en lesiones hiperqueratósicas que afectaban nudillos y rodillas. Presentaba una deformidad de los uñas de las manos, de aspecto de pico de loro, acompañada de dolor y moderada hiperhidrosis. Se llegó al diagnóstico de queratodermia ortoqueratósica difusa de Unna Thost. Esta queratodermia se presenta en una de cada 40 000 personas y es de origen hereditario. Por lo infrecuente de su presentación, la afectación biológica, repercusión psicológica y limitación laboral que presupone, así como el seguimiento médico que lleva, se decidió presentar este caso.
Palabras clave: queratodermia palmoplantar.
ABSTRACT
The case of a 37 years old, black skinned, male patient who attended Dermatology Consultation in his health area is presented. The patient had skin lesions on the palms of his hands and the soles of his feet. He stated that these injuries had worsened with time. Eventually they became hyperkeratotic lesions affecting the knuckles and the knees. The patient also presented a deformity of his fingernails resembling a parrot’s beak that produced pain and moderate hyperhidrosis. A diagnosis of diffuse orthokeratotic Thost-Unna keratoderma was completed. This keratoderma occurs in one out of every 40 000 people and is hereditary. Because it is such an uncommon presentation with biological implications, psychological impact and work impediments, that requires a specific medical monitoring, it was decided to present this case to be published.
Key words: keratoderma, palmoplantar.
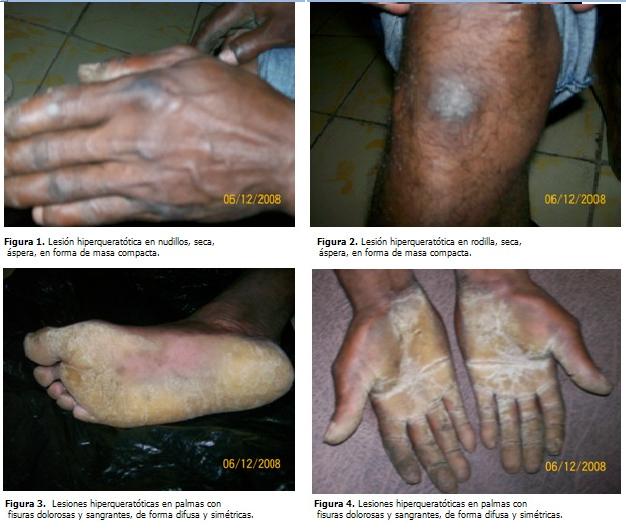
INTRODUCCIÓN Las queratodermias hereditarias incluyen: Existe otra clasificación que las divide en: Las queratodermias simples difusas comprenden QPP: La queratodermia palmo plantar de Unna Thost o queratodermia ortoqueratósica difusa como también se le conoce, es una de las queratodermias hereditarias que con mayor frecuencia se presenta en la práctica dermatológica. La incidencia es de un caso cada 40 000 personas. Se hereda como un trastorno autosómico dominante. Aparece durante los dos primeros años del nacimiento y perdura toda la vida. (1,2,5) Clínicamente, la enfermedad se caracteriza por una queratodermia bien delimitada, simétrica y a menudo cerúlea que involucra a toda la región palmo plantar y se interrumpe bruscamente en el nivel de las muñecas, con una banda eritematosa en la periferia. (6-9) La propagación de la hiperqueratosis hacia el dorso de las manos y de las muñecas es un rasgo variable en la mayoría de las familias. A menudo la enfermedad afecta el dorso de las manos con un engrosamiento símil- esclerodermia de la piel distal con respecto a la articulación intrefalángica proximal. Hay frecuente hiperhidrosis y dolor en las zonas que se fisuran. No hay cambios asociados en el cabello y/o dientes. (10) El aspecto en pico de loro de las uñas de las manos y el ensanchamiento de la banda onicorneana son hallazgos característicos de esta enfermedad. Se puede observar una hiperqueratosis de los nudillos en empedrado, pero el trastorno afecta rara vez los codos y las rodillas. La infección secundaria por dermatofitos, con maceración y exfoliación de palmas y plantas, es una complicación frecuente de esta variante de queratodermia. (6,11) La histología de este patrón muestra hiperqueratosis marcada, con acantosis y papilomatosis, se diferencia de la QPP epidermolítica por la ausencia de epidermólisis. La evaluación histológica de miembros de la familia original con la enfermedad de Unna Thost originó dudas acerca de la existencia real de este patrón de QPP y llevó a investigadores posteriores a considerar que esta enfermedad es una identidad clínica independiente. (12,13) En su tratamiento se deben tener en cuenta: En presencia de una dermatofitosis coexistente es eficaz un tratamiento prolongado con un antimicótico sistémico; por ejemplo, el uso del itraconazol en dosis de 100 mg al día. La pomada de Whitfield es un agente queratolítico tópico eficaz. Son enfermedades crónicas que no mejoran con el transcurso de la vida, en algunas formas clínicas los pacientes quedan inutilizados. (1-5, 8 ,11-13) PRESENTACIÓN DEL CASO En los antecedentes patológicos familiares se recogió: madre, hermana y hermano materno con discreta hiperqueratosis, tres tíos y cuatro tías por vía materna con lesiones similares, las cuales desaparecieron al arribar a la tercera edad. Sus tres hijos no presentaban lesiones. La piel presentaba lesiones hiperqueratósicas en palmas y plantas, con fisuras dolorosas y sangrantes de forma difusa y simétrica, rodeadas de halo eritematoso. Lesión hiperqueratósica seca, áspera en forma de masa compacta en rodillas y nudillos. (Figuras 1 a 4).
Las queratodermias pertenecen al grupo de las genodematosis y son aquellas dermatopatías heterogéneas cuyas principales manifestaciones se presentan en piel y anejos; tienen como elemento común su condicionamiento genético. Se produce por trastornos de la queratinización junto a la ictiosis, pitiriasis rubra pilaris y la poroqueratosis de Mibelli. (1-4)
Estas afecciones se distinguen por un engrosamiento anormal de palmas de las manos y plantas de los pies. Se clasifican en dos grandes grupos: hereditarias y adquiridas.
Para el tratamiento sistémico se emplean los derivados retinoicos como el etretinate de 1-1,5 mg /Kg. /día.
Se presenta el caso de un paciente de color de piel negra, masculino, de 37 años de edad, jubilado, que desde los 2 años de edad, según refirió, la madre comenzó a notarle aspereza en las manos. Durante sus estudios en el Politécnico Forestal de Pinar del Río presentó un incremento de los síntomas que fue interpretado como alergia a las plantas. Posteriormente trabajó en la refinería de petróleo donde manifestó incapacidad para realizar sus activididades laborales por lo que acudió a la consulta de dermatología de su área de salud.
A la exploración física se observó lo siguiente:
Uñas deformadas con aspecto en pico de loro, con conservación de su aspecto blanco. Ensanchamiento de repliegues supraungulares. (Figura 5).

No apareció ninguna otra alteración cutánea.
Exámenes complementarios:
Se le realizó biopsia de piel. El examen histológico reveló hiperqueratosis marcada, acantosis y papilomatosis con ausencia de epidermólisis de los queratinocitos de la capa basal y granulosa compatible con queratodermia ortoqueratósica de Unna -Thost.
CONCLUSIONES
Después de corroborar la clínica con el estudio histopatológico se pudo concluir que el paciente es portador de una queratodermia ortoqueratósica de Unna-Thost, afección heredo –familiar que por lo infrecuente de su presentación, la afectación biológica, repercusión psicológica y limitación laboral que presupone es elemento importante a incorporar al arsenal de conocimientos de nuestros profesionales.
El paciente que ahora se presenta, se mantiene en consulta para realizar el control y seguimiento de sus manifestaciones clínicas con el adecuado apoyo emocional necesario en esta entidad dermatológica.
REFERENCIAS BIBLIOGRÁFICAS
1. Falcón Lincheta LJ. Genodermatosis. En: Manssur J, Díaz Almeida J, Cortés Hernández M. Dermatología. La Habana: Editorial Ciencias Médicas; 2002: p. 53-57
2. Domonkos AN. Algunas genodermatosis y síndromes adquiridos. Ciudad de La Habana: Editorial Científico-Técnica; 2004
3. Caputo R, Tadini G. Striate keratoderma. En: Atlas Ofgenodermat. London: Taylor & Francis; 2006: p. 84
4. McRonald F, Liloglou T, Xinarianos G, Hill L, Rowbottom L, Langan J, et al. Down-regulation of the cytoglobin gene, located on 17q25, in tylosis with oesophageal cancer (TOC): evidence for trans-allele repression. Human Molecular Genetics. 2006;15(8):1271-77
5. Wan H, Dopping-Hepenstal PJ, Gratian MJ, Stone MF, Zhu G, Purkis PE, et al. Striate palmoplantar keratoderma arising from desmoplakin and desmoglein 1 mutations is associated with contrasting perturbations of desmosomes and the keratin filament network. Br J Dermatol. 2004;150:878-91
6. Pastor MA, González L, Kilmurray L, Bautista P, López A, Puig AM. Queratodermia acuagénica: tres nuevos casos y revisión de la literatura. Actas Dermosifiliogr. 2008;99:399-406
7. Yaghoobi R, Omidian M, Sina N, Abtahian S, Panahi MR. Olmsted Syndrome in an Iranian Family: Report of Two New Cases. Arch Iranian Med. 2007;10(2):246-9
8. Xiang-Qun G, Qing Sh, Lounsbury S, Bai D, Laird D. Functional Characterization of a GJA1 Frameshift mutation Causing Oculodentodigital Dysplasia and Palmoplantar Keratoderma. J Biol Chem. 2006;281(42):31801-11
9. Mallory SB. Palmoplantar keratoderma. En: An Illustrated dictionary of dermatologic syndromd. 2th. ed. London: Taylor & Francis; 2006: p. 191
10. Shah J, Goel S. Papillon-Lefevre syndrome: Two case reports. Indian J Dent Res [revista en Internet]. 2007 [citado 12 May 2010];18:[aprox. 3p]. Disponible en: http://www.ijdr.in/text.asp?2007/18/4/210/35834
11. de Haar S, Hiemstra P, van Steenbergen T, Everts V, Beertsen W. Role of Polymorphonuclear Leykocyte-Derived serine proteinases in defense against Actinobacillus actinomycetemcomitans. Infect Immun. 2006;74(9):5284-91
12. Bertrand F, Plantard L, Aeschbacg L, Brakch N, Christen Zaech S, Viragh P, et al. SLURP1 Is a Late Marker of Epidermal Differentiation and Is Absent in Mal de Meleda. Journal of Investigative Dermatology. 2007;127:301-308
13. Padial A, Morales V, Armario-Hita JC, Ingunza JL, Fernández Vozmediano JM. Queratodermia palmoplantar varians (striata et areata) tipo acroqueratosis esencial crónica de Degos. Actas Dermosifiliogr. 2008;99:134-7
Recibido: 20 de julio de 2011.
Aprobado: 20 de julio de 2011.
Graciela Caridad Cabrera Acea. Especialista de II Grado en Dermatología. MSc. en Enfermedades Infecciosas. Profesor Instructor. Policlínico Laboral Universitario. Área 7. Cienfuegos. Correo electrónico: medcgc900831@fcmc.cfg.sld.cu


{kind=link}