










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkMediSur
versión On-line ISSN 1727-897X
Medisur vol.16 no.2 Cienfuegos mar.-abr. 2018
PRESENTACIÓN DE CASO
Feocromocitoma quístico. Presentación de un caso
Cystic Pheochromocytoma. Case presentation
Javier Martínez Navarro , Lisanka Fumero Roldán , Erika Izquierdo Reyes , Martha Yudey Rodríguez Pino
Hospital General Universitario Dr. Gustavo Aldereguia Lima, Cienfuegos, Cuba
RESUMEN
Los feocromocitomas son tumores relativamente raros. Constituyen lesiones hipervascularizadas que, en un 90% de los casos, producen hipertensión arterial. Generalmente reportadas como lesiones sólidas, debido a su hipervascularización, pueden presentar áreas con degeneración quística focal o parcial. Sin embargo, aunque infrecuentes, se han reportado feocromocitomas con degeneración quística total o subtotal y son denominados feocromocitomas quísticos. Por tales razones se decidió la publicación del caso de un paciente masculino que falleció en el Hospital Gustavo Aldereguía Lima, a los 65 años de edad, con historia personal de hipertensión arterial de 10 años de evolución, artritis gotosa desde hacía cinco años y diabetes mellitus, todas bajo tratamiento regular. Acudió a consulta por cefalea universal y náuseas. Estudios imagenológicos evidenciaron una masa tumoral en glándula suprarrenal derecha. Durante su ingreso presentó edema pulmonar severo no cardiogénico que le causó la muerte. El estudio necrósico confirmó que presentaba un feocromocitoma quístico.
Palabras clave: feocromocitoma, informe de casos.
ABSTRACT
Pheocromocytomas are relatively unusual tumors. They are vascularized lesions that, in the 90% of the cases produce arterial hypertension. Generally reported as solid lesions due to hyper-vascularization. They may present areas with focal or partial cystic degeneration. Although infrequent, however, pheochromocytomas with total of subtotal cystic degeneration has been reported and are called cystic Pheochromocytomas. Due to these reasons it was decided to publish the case of a male patient who died at the Gustavo Aldereguía Lima Hospital, aged 65 with a ten year personal history of arterial hypertension, gout arthritis and nausea. He came to the hospital complaining of universal cephalea and nausea. Imaging studies showed a tumorous mass in the suprarenal right gland. During admission he presented a non-cardiogenic severe pulmonary edema which caused his death. The necropsy study confirmed a cystic pheochromocytoma.
Key words: pheocromocitoma, case report.
INTRODUCCIÓN
Los feocromocitomas (F) son tumores relativamente raros, se observan aproximadamente en el 0,1 % de la población de sujetos hipertensos. Se origina en las células cromafines de la médula suprarrenal (MS), que produce, almacena y secreta catecolaminas en exceso (adrenalina, noradrenalina, dopamina). Estas hormonas son las causantes del cuadro clínico que presentan los pacientes cuyo síntoma principal es la hipertensión arterial (HTA) que puede ser sostenida o paroxística y en ocasiones grave.1 Estos tumores pueden originarse también a partir de las células cromafines situadas en los ganglios simpáticos o en sus proximidades. En el 85 % de los casos se localizan en la MS y el 15 % restante son extrasuprarrenales. La distinción patológica entre los feocromocitomas benignos y malignos no está muy clara, la malignidad oscila entre 5-46 %.2 El diagnóstico del F se realiza por medio de la clínica, el laboratorio y la imagenología.3 Los feocromocitomas quísticos (FQ) constituyen lesiones adrenales infrecuentes. Existen escasos reportes en la literatura mundial de FQ parciales o totales que describen la apariencia de estas lesiones, por lo tanto, el propósito de este trabajo es reportar un raro caso de FQ en un paciente de 65 años de edad, cuyo diagnóstico definitivo se estableció tras practicar la autopsia clínica (AC). Esta entidad, por su frecuencia de presentación y el escaso material bibliográfico disponible, merece su comunicación.
PRESENTACIÓN DEL CASO
Paciente masculino, de 65 años de edad, de procedencia rural y con antecedentes patológicos personales de obesidad, HTA y cardiopatía isquémica de 10 años de evolución para lo cual consumía de forma regular enalapril a dosis de 20 miligramos (mg) al dia, nitropental 20 mg cada ocho horas. Artritis gotosa desde hacía cinco años para lo cual cumplía tratamiento regular con alopurinol, 100 mg cada ocho horas. Diabetes mellitus de cuatro años de evolución tratada con metformina 500 mg tres veces al dia. Acudió a su médico de asistencia por cefalea universal de intensidad moderada y náuseas ocasionales. Datos positivos al examen físico: Peso: 95 Kilogramos. Talla: 1,65 metros Índice de masa corporal: 34,8 Aparato cardiovascular: ruidos cardiacos taquicárdicos, de buen tono e intensidad. Frecuencia cardíaca: 115 latidos por minutos. Presión arterial 160/110 mm Hg. Se indicaron estudios complementarios e imagenológicos de forma ambulatoria cuyos resultados fueron: Hemoglobina: 120 g/L Hematocrito: 0,39 Eritrosedimentación 19 mm/h Leucocitos totales: 8.0 X 109 L Segmentados: 52 % Eosinófilos: 2 % Linfocitos: 46 % Hemoglobina glicosilada: 6,2 % Glicemia: 9,44 mmol/L Creatinina: 87 umol/L Urea: 465 umol/L Ácido úrico: 704 umol/L Colesterol: 4,80 mmol /L Triglicéridos: 1,29 mmol/L Transaminasa glutámico pirúvica: 20 U/L Transaminasa glutámico oxalacética: 6 U/L Lactodeshidogenasa: 289 U/L Fosfatasa alcalina: 178 U/L Proteínas totales: 75 g/L Globulina: 33 g/L Serina: 42 g/L. Eletrocardiograma: taquicardia sinusal. FC: 110 latidos por minuto. Ultrasonido (UTS) abdominal: hígado de tamaño normal con marcado aumento de su ecopatrón de aspecto graso. Vesícula biliar con imágenes de litiasis de 4 y 8 mm y de paredes finas. Vías biliares y páncreas normales. Riñón izquierdo (RI) de tamaño normal con quiste de 12 mm hacia el polo inferior que no produce hidronefrosis. Riñón derecho que impresiona de tamaño normal, observando hacia su polo superior una imagen predominantemente quística de 56 X 65 mm y que pudiera estar comprometiendo la glándula suprarrenal de ese lado. Litiasis en el grupo calicial medio. Vejiga normal. Se sugiere tomografía axial computarizada de abdomen (TAC). (Figura 1). El paciente comenzó con dolores articulares de intensidad moderada en el pie derecho a nivel del tobillo, acompañado de enrojecimiento y aumento de volumen. Estos signos y síntomas fueron interpretados como un ataque agudo de artritis gotosa. Se ingresó en el Servicio de Medicina Interna del hospital donde se le indicaron analgésicos, antiinflamatorios no esteroideos y alopurinol 400 mg al día. Se indicaron otros exámenes de laboratorio y se realizó la TAC de abdomen. Los nuevos estudios analíticos y TAC indicados informaron: Cortisol basal: 229,7 mmol/L Cortisol postinihibición con 2 mg de dexametasona: 15,96 mmol/L Insulinemia basal: 4,69 mmol /L Ácido vanililmandelico en orina en 24 horas: negativo. Tomografía axial computarizada de abdomen: se realizó estudio simple y endovenoso (EV) con el paciente en ayunas. Se observó aumento de tamaño de la glándula suprarrenal derecha que mide 43 x 36 mm con centro más hipodenso (densidad líquida) y halo hiperdenso que no aumenta su densidad después de la administración de contraste EV en relación con presencia tumoral. (Figura 2). El paciente comenzó de forma súbita con disnea intensa que llegó a impedir el decúbito. Este cuadro se interpretó nosológicamente como un edema agudo del pulmón, del cual no logró estabilizarse y el paciente falleció. Se autorizó y practicó la AC donde se constató edema pulmonar severo no cardiogénico (causa directa de muerte) y F con degeneración quística parcial de glándula suprarrenal derecha. Otros hallazgos en la autopsia fueron: colecistitis crónica litiásica, esteatosis hepática macrovacuolar severa, ateromatosis moderada de la aorta y sus ramas, quiste simple en polo inferior de RI, gastritis crónica moderada antral, duodenitis crónica moderada y congestión visceral generalizada.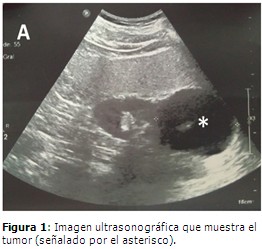

DISCUSIÓN
Los síntomas del F son secundarios a la hipersecreción de catecolaminas y su efecto en diferentes órganos. La triada de síntomas clásicos de esta enfermedad (cefalea, diaforesis y palpitaciones) tiene pobre sensibilidad y especificidad, se encuentran solamente en el 30 % de los pacientes. Otros síntomas descritos son: ansiedad, pánico, disnea, epigastralgia, dolor torácico, náusea, vómito, palidez, temblor y manos frías. La HTA puede ser episódica, continua o una presión arterial normal. Los ataques catecolaminérgicos son episódicos con una duración de 15 a 20 minutos y pueden presentarse varias veces al día. Pueden iniciar con una sensación de calor en cara y en tórax que progresa a palpitaciones, cefalea, temblor, palidez y diaforesis. Las manos se encuentran frías, lo mismo que los pies y puede existir fenómeno de Raynaud. Algunos casos pueden presentar visión borrosa, papiledema, pérdida de peso, poliuria y polidipsia. Raramente se asocia con cardiomiopatía, atribuida a un exceso de catecolaminas, la cual es similar a la cardiomiopatía inducida por el estrés. Los pacientes pueden presentar edema pulmonar.5 El paciente cuyo caso se presenta, sufrió de HTA durante diez años y la mantuvo controlada con tratamiento medicamentoso. En las últimas semanas mantuvo hipertensión continua y refractaria presentando además cefalea y náuseas. Fallece a consecuencia de un episodio de disnea intensa secundario a edema pulmonar masivo. El caso que se reporta sobrepasa los 60 años de edad. La literatura reporta que estos tumores se presentan con mayor frecuencia entre la tercera y cuarta década de vida. La alta mortalidad que implica, y el hecho de que en su mayoría sean curables quirúrgicamente, hacen que sea imprescindible un diagnóstico precoz,6 el cual no pudo realizarse en nuestro paciente debido a su fallecimiento imprevisto. En los adultos, 80 % de los estos tumores son unilaterales y solitarios, como en este caso. Pueden alcanzar un gran tamaño, pero la mayor parte de ellos pesa menos de 100 gramos y mide menos de 10 cm.7 coincidiendo esto con las dimensiones del tumor reportado. (Figura 3). Tienen predominio en el lado derecho, sin conocerse la causa. En sentido general, el 90 % de estas neoplasias cromafines tienen una presentación esporádica, frente a solo un 10 % con presentación familiar. En este último caso se sabe que puede heredarse de forma aislada con herencia autosómica dominante o dentro de síndromes hereditarios, tales como: enfermedad de Von Hippel Lindau, enfermedad de Von Recklinghausen, la enfermedad de Sturge Weber y la esclerosis tuberosa.8 Este artículo presenta un caso esporádico, desarrollado en la glándula suprarrenal derecha. Los estudios histológicos de este caso arrojaron resultandos en congruencia con el feocromocitoma. (Figura 4). Estos tumores presentan frecuentemente degeneración quística parcial o focal debido a su hipervascularización. Sin embargo, en ocasiones, pueden sufrir degeneración quística total o parcial -como lo observado en este paciente-, aunque este tipo de presentación resulta ser muy infrecuente. Si bien el proceso fisiopatológico de la degeneración quística no se encuentra aun perfectamente aclarado, se ha postulado que la misma sería secundaria a hemorragia intratumoral con posterior necrosis y reabsorción, siendo el componente quístico del F consecuencia de necrosis y licuefacción intratumoral. Se consideraría como mecanismo “gatillo” el rápido crecimiento del tumor asociado a insuficiente aporte vascular, lo cual sucedió en el presente caso. El FQ suele ser observado en ecografía como una lesión sólida con múltiples áreas de aspecto quístico en su interior. En TAC, puede manifestarse el componente sólido asociado a múltiples áreas necrótico-quísticas, que demuestran baja atenuación.9Estas características fueron observadas en los estudios imagenológicas practicados al paciente que se analiza. Ante la sospecha clínica de F, el paso inicial para el diagnóstico es la confirmación bioquímica de la producción excesiva de catecolaminas. Las pruebas bioquímicas disponibles incluyen medición de catecolaminas plasmáticas y urinarias, metanefrinas (metanefrina y normetanefrina) plasmáticas y urinarias y ácido vanililmandélico (VMA) en orina.10 En relación a esto, en nuestro caso solo se logró realizar la dosificación de VMA en orina la cual resultó negativa.

REFERENCIAS BIBLIOGRÁFICAS
1. Santos Monzón Y, Plain Pozos C, Pérez de Alejo Alemán A. Feocromocitoma. Presentación de un caso. Medisur [revista en Internet]. 2014 [citado 7 Ago 2017];12(4):[aprox. 2p]. Disponible en: http://www.medisur.sld.cu/index.php/medisur/article/view/18871
2. Piriz Momblant A, Vázquez Vilanora R. Feocromocitoma y neurofibromatosis: una rara asociación. Revisión del tema a propósito de un caso. Rev Inf Cient [revista en Internet]. 2017 [citado 7 Ago 2017];96(4):[aprox. 16p]. Disponible en: http://revinfcientifica.sld.cu/index.php/ric/article/view/1197
3. Uribe J, González M, Tamayo L. Manifestaciones inusuales del feocromocitoma: Reporte de caso y revisión de la literatura. Rev Colomb Cardiol [revista en Internet]. 2016 [citado 7 Ago 2017];23(2):[aprox. 5p]. Disponible en: http://www.scielo.org.co/scielo.php?script=sci_arttext&pid=S0120-56332016000200013&lng=en
4. Ahualli J, Méndez Uriburu L, Ravera María L, Méndez Uriburu M. Feocromocitoma quístico: a propósito de un caso. Rev Argent Radiol [revista en Internet]. 2008 [citado 7 Ago 2017];72(2):[aprox. 4p]. Disponible en: http://www.scielo.org.ar/scielo.php?script=sci_arttext&pid=S1852-99922008000200009&lng=es
5. Román González A, Builes Barrera CA, Gutiérrez J. El primer caso de feocromocitoma. Rev Col Endocrinol Diab Metab [revista en Internet]. 2016 [citado 7 Ago 2017];3(2):[aprox. 4p]. Disponible en: http://www.revistaendocrino.org/index.php/rcedm/article/view/33
6. González González JL, Menéndez Núñez J, Escarpanter JC, Lazo Alba A. Miocardiopatía dilatada y feocromocitoma. Rev Cubana Cir [revista en Internet]. 2013 [citado 7 Ago 2017];52(1):[aprox. 7p]. Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0034-74932013000100007&lng=es
7. Hernández Hernández G, Jasso Barranco R, Lozano Corona R, Jaramillo Martínez C. Feocromocitoma sin manifestaciones clínicas y bioquímicas, resecado por vía laparoscópica, reporte del caso y revisión de la literatura. Rev Mex Cir Endoscóp [revista en Internet]. 2013 [citado 7 Ago 2017];14(1):[aprox. 4p]. Disponible en: http://new.medigraphic.com/cgi-bin/resumen.cgi?IDARTICULO=47455
8. Miranda Folch JJ, García Cuervo D, Vega Jiménez J, Alemán Marichal B, Jiménez Álvarez A, Castelnau Sánchez A. Arterial hypertension secondary to sporadic pheochromocytoma. Case presentation. Rev Med Electrón [revista en Internet]. 2016 [citado 7 Ago 2017];38(1):[aprox. 10p]. Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S1684-18242016000100008&lng=es
9. Takarabe D, Takahashi Y, Tsujimoto T, Noto H, Kishimoto M, Minowada S, Noda M. Cystic Pheochromocytoma Discovered as an Adrenal Incidentaloma. J Med Cases [revista en Internet]. 2013 [citado 7 Ago 2017];4(11):[aprox. 5p]. Disponible en: http://www.journalmc.org/index.php/JMC/article/view/1594
10. Enríquez A, Paredes A, Tagle R, Castro P. Feocromocitoma: presentación como síndrome coronario agudo: Reporte de un caso. Rev Méd Chile [revista en Internet]. 2014 [citado 7 Ago 2017];142(2):[aprox. 5p]. Disponible en: http://www.scielo.cl/scielo.php?script=sci_arttext&pid=S0034-98872014000200015&lng=es
Recibido: 21 de noviembre de 2017.
Aprobado: 06 de abril de 2018.
Javier Martínez Navarro. Especialista de I Grado en Anatomía Patológica. Hospital General Universitario Dr. Gustavo Aldereguia Lima. Cienfuegos. Correo electrónico: javiermn@jagua.cfg.sld.cu

