










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Habanera de Ciencias Médicas
versión On-line ISSN 1729-519X
Rev haban cienc méd v.7 n.1 Ciudad de La Habana ene.-mar. 2008
Centro de Referencia Nacional de Retinosis Pigmentaria. (CRNRP)
Hospital Docente Dr. Salvador Allende.
RETINOSIS PIGMENTARIA: CLÍNICA, GENÉTICA Y EPIDEMIOLOGÍA EN ESTUDIO DE FAMILIAS HABANERAS.
** Raisa Hernández Baguer. Calle 11 núm. 2722 entre Reforma y Canal. Rpto. Antonio Maceo Cerro. Teléfono:878-8163. raisa.baguer@infomed.sld.cu
** Mirta Copello Noblet. Hernán Ben 2119 entre Kessel y San Leonardo. Víbora Park. Arroyo. Teléfono: 644-8944.mirtacopello@infomed.sld.cu
*** Beatriz Dyce Gordon. Cintra núm.66 entre Reyes e Infanta. Cerro. bgordon@infomed.sld.cu
* Moraima Rodríguez Alba. Loma y Tulipán. Edificio ICP. Apto.30 Plaza. Teléfono: 881-7406. moralba@infomed.sld.cu
* Angel Arce Alvarez. Estrada Palma núm. 555 entre Mayía Rodríguez y Goicuría. Santo Suárez, 10 de Octubre. Ciudad de La Habana. Teléfono: 641-8916. angelcustodioa@infomed.sld.cu
**Georgina Saint- Blancard Morgado. Ayuntamiento núm.79 entre Conill y Tulipán. Plaza. Ciudad de La Habana. Teléfono: 870-0388. cachitagsb@yahoo.es
* Fructuoso Suárez Herrera. Calle 16 núm. 467 entre Dolores y Concepción. 10 de Octubre. Ciudad de La Habana. Teléfono: 69-5579
*Julián Pastor Pérez Lara. Lawton núm.1510 entre San Mariano y Vista Alegre. 10 de Octubre. Ciudad de La Habana.
*Especialista Primer Grado en Oftalmología.
**Especialista Segundo Grado en Oftalmología.
***Especialista Primer Grado en Genética Clínica.
Centro de Referencia Nacional de Retinosis Pigmentaria. (CRNRP) Hospital Docente Dr. Salvador Allende.Calzada del Cerro 1551, Cerro. Ciudad de La Habana, Cuba. Telef.877-6358
RESUMEN
Se realiza una investigación descriptiva, retrospectiva y transversal con el objetivo de conocer los resultados de la pesquisa, clasificación y abordaje multidisciplinario de la retinosis pigmentaria (RP) en Ciudad de La Habana en los años 2006-2007. Se analizan las historias clínicas y encuestas individualesde cada paciente así como las fichas y árboles genealógicos de sus familias diseñadas por el Programa Nacional de Retinosis Pigmentariapara establecer las características clínicas, genéticas y epidemiológicas de la RP en ellos. Se hace el resumen porcentual de los resultados que se muestran en gráficos.
Palabras clave: retinosis pigmentaria, herencia, familia, ceguera nocturna.
INTRODUCCIÓN
En nuestro país habitan 46,455 discapacitados visuales de los cuales aproximadamente 4,123 están aquejados de Retinosis Pigmentaria (RP) para una tasa nacional de 4,1x 10,000 habitantes. (1, 2)
Esta enfermedad de origen genético y comportamiento familiar
es la primera causa de déficit visual importante dentro de las causas distróficas y degenerativas oculares. Ocasiona como síntoma inicial ceguera nocturna o fotofobia según el tipo clínico de que se trate, lo cual empeora en el transcurso de la vida afectando progresivamentela agudeza visual periférica, luego la visión central y el campo visual así como el fondo de ojo. En otros casos se afecta primero la lectura y la visión de colores.(3,4, 5)
La evolución de la enfermedad determina varias etapas señaladas como incipiente, de estado, avanzada y muy avanzada que hemos marcado como Estadios I, II, III y IV respectivamente en la clasificación cubana que nos propusieran como instrumento de los oftalmólogos cubanosel Dr. CM Profesor Dr. Orfilio Peláez Molina (†) y colaboradores. (6)
El hecho de estudiar una enfermedad hereditaria como la RP teniendo como unidad básica la familia constituye un noble propósito que se inserta dentro de la Medicina Preventiva y Familiar de nuestra política de salud, con el empeño de elevar cuantitativa y cualitativamente el diagnóstico precoz de esta enfermedad en la comunidad que puede llevar a la discapacidad visual. (7, 8)
Por la heterogeneidad genética y clínica de la RP, ésta puede presentarse aislada (no sindrómica) o formando parte de síndromes (sindrómica).
La caracterización clínica, genética y epidemiológica de la enfermedad en nuestra provincia es un motivo constante de investigación dentro de nuestro programa de trabajo dedicado a atender esta distrofia ocular, con el objetivo de clasificar las familias aquejadas teniendo en cuenta aspectos que permitan conocer el estadio evolutivo de la enfermedad en los afectados, el grado de salud visual residual y facilitar la consiguiente realización de investigaciones o la aplicación de terapéuticas novedosas, buscando prolongar la calidad de vida de nuestros pacientescon un apropiado balance entre discapacidad y rehabilitación.
MATERIAL Y METODO
Se tomaron las encuestas individuales diseñadas por el Programa Nacional de Retinosis Pigmentaria y las historias clínicas, fichas familiares y árboles genealógicos de cada afectado previo consentimiento informado haciéndoles saber que ningún proceder que se les practicase iba en contra de los principios de la Salud Pública en Cuba ni en detrimento de la suya en particular y que se podían retirar de la investigación si lo deseaban. Se realizó el registro de variables como: edad y género, tipo de herencia, examen oftalmológico completo (comprendiendo fondo de ojo, campo visual, electrorretinograma (ERG) y agudeza visual), edad de debut de los primeros síntomas, presencia de consanguinidad familiar, de otras enfermedades oculares y generales asociadas para realizar la clasificación clínica de la RP , del estadio evolutivo actual de cada enfermo (6) y del riesgo familiar de aparición de nuevos casos aún no diagnosticados. Tras el vaciamiento de datos y el análisis de los mismos, se muestran los resultados de forma porcentual utilizando gráficos y cuadros.
Operacionalizacion de las variables
Edad: Tomada en años cumplidos de acuerdo a la declaración del entrevistado agrupada en intervalo de 10 años hasta 89 años para facilitar su comparación con estadísticas internacionales.
Género: Masculino Femenino
Edad de debut: Edad en que aparecen los primeros síntomas
Antecedentes personales y familiares: Solamente aquellos que son considerados de importancia y con + de 5 años de evolución
Otras enfermedades oculares: Catarata, glaucoma, ametropia. Registrar la entidad según su nombre propio.
Consanguinidad: Parentesco entre personas que descienden del mismo tronco.
Clasificación Clínico-evolutiva de la RP: Agudeza visualCampo visual, Fondo de ojo (Ver Anexo)
Clasificación del Tipo de herencia: Según definición de Mc Kusick y Kaplan
Familia: Parentela unida por lazos de sangre
RESULTADOS
Se estudiaron en Ciudad de La Habana 613 pacientes de ambos sexos, aunque predominó el género masculino, con diagnóstico confirmado de RP por electrorretinograma alcanzando la provincia una tasa de prevalencia de 2,7 x 10,000 habitantes teniendo en cuenta el total de la población habanera en el momento del estudio.El rango de edades para todos abarcó de 10 a 75 años, sobresaliendo el grupo de 61 años y más, con 190 enfermos (39,3%), seguido por el de 31 a 40 años grupo que está laboralmente activo con 135 pacientes (27.9%).
Los 613 pacientes estaban distribuidos en 483 familias de las cuales 214 proceden de otras provincias del país, aunque varios de sus miembros residen actualmente en la nuestra.
La edad de comienzo de la ceguera nocturna fue después de los 20 años en 187 familias (38,7%) con uno o varios miembros afectados marcando un tipo de RP de comienzo tardío en su debut después de terminada la adolescencia. (2).
La consanguinidad que generalmente está presente en el tipo de herencia autosómica recesiva estuvo presente en 49 familias (10,1%) lo que se corresponde con la poca información que tenía la población en el siglo pasado en cuanto a este factor de riesgo para la aparición de algunas enfermedades en la descendencia.
DISCUSIÓN
Al incluirse entre los tipos de herencia de la RPel recesivo ligado al cromosoma X donde los afectados siempre son varones, en nuestra población total de enfermos predomina el sexo masculino como ocurre en otros países del mundo. (2, 3, 4, 5)
La herencia autosómica recesiva se expresa de forma prioritaria enlos árboles genealógicos, pero aún predominan los enfermos que
no tienen herencia determinada (casos simples o múltiples) 242 pacientes para un 50,3% loque indica alta tasa de nuevas mutaciones. Gráfico. 1.
Esto se manifiesta igualmente en Estados Unidos, Francia, Japón y Dinamarca. (2, 5, 9).
Se impone entonces en nuestra labor de prevención y promoción de saludel estudio de estas familias sin herencia conocida hasta el momento, para el diagnóstico precoz de otros probables enfermos que aún estén en la etapa presintomática de la ceguera nocturna.
Las características fondoscópicas determinaron que la tipicidad del cuadro clínico se observó en 424 familias (87,7%) independiente
de la herencia. (10).La atipicidad en los casos fue de tipo sectorial, central y monocular en el fondo de ojo y estuvo presente en 16 familias (12,3%) (2)
La RP sindrómica se presentó en 43 familias (8,8%) con 51 pacientes. Este resultadose muestra en el Gráfico 2
Se destacó de forma preferente el Síndrome Usher I y II entre otros. (Bardet-Bield, Hunter y Hurler) (11, 12). En su cuadro clínico yevoluciónesta RP asociada se comporta como típica en cuanto a la distribución de los pigmentos en el fondo lo que fue hallado en el análisis individual del examen oftalmológico de cada enfermo. La evolución de la enfermedad se presenta en estadios avanzado y muy avanzado en 152 familias y 130 familias respectivamente con Retinosis Pigmentaria no sindrómica, no precisandose bien el tipo de evolución en 22 familias para un 4,6%. Gráfico 3.
Teniendo en cuenta que en algunas familias el debut de la enfermedadfue antes de los 20 años y la tendencia nacional actual en Cuba y enAmérica Latina y es tener una población cada vez más longeva, es posible que en algunos afectados se llegue a observar la enfermedad hasta llegar a esos estadios. (12, 13)
Queremos destacar que no siempre los enfermos de las familias con RP recesiva ligada al cromosoma X en nuestro estudio han tenido lapeor evolución hacia la ceguera como expresa la literatura revisada y publicada hasta el momento. (2, 3, 4)
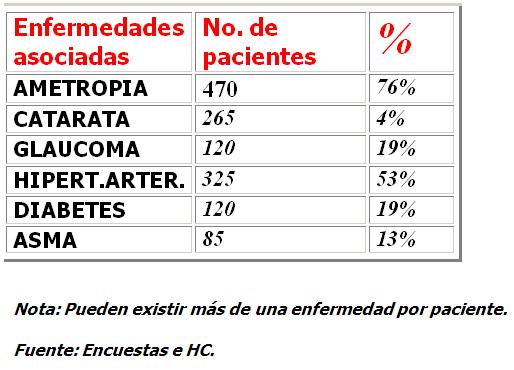
Las otras enfermedades oculares que más frecuentemente se asocian a la RP son ametropias, catarata y glaucoma cuyo tratamiento debe acompañar al de la RP. Las enfermedades generales registradas como antecedentes patológicos personales y familiares que se señalan con mayor frecuencia entre otras son la hipertensión arterial, la diabetes mellitus y el asma bronquial en ese orden deaparición. Tabla 1.
Consideramos importante conocer el estado de salud individual y familiar y la concomitancia de otras enfermedades crónicas no transmisibles que requieren de una intervención médica y seguimiento apropiados en estas familias, teniendo en cuenta si fuera necesario la intersectorialidad para asegurar y favorecer un buen estado de salud general, una destacada participación social y calidad de vida satisfactorios a pesar del padecimiento crónico ocular.
CONCLUSIONES
1-Las características clínicas, genéticas y epidemiológicas de pacientes y familias con Retinosis Pigmentaria (RP) en Ciudad de La Habana se comportó de forma semejante a las descritas en estudios realizados en otras provincias cubanas y en las principales ciudades de países desarrollados. No son confiables registros de países con menos desarrollo socioeconómico que hemos consultado para este trabajo.
2-Predominó el tipo clínico de RP con características fondoscópicastípicas y la herencia autosómica recesiva.
3-El grado de progresión (estadio clínico) de la enfermedad es extremadamente variable de una familia a otra e independiente del tipo de Retinosis Pigmentaria y dentro del mismo tipode herencia.
4-La Prevalencia por grupo de edadesfue predominante en latercera edad lo quenos confirma que en la actual sociedad cubana el adulto mayorocupa una cifra importante dentro de nuestra población pugnando por una longevidad satisfactoria.
RECOMENDACIONES
Es necesario establecer el enfoque familiar en el estudio de esta enfermedad de origen genético para una deteccción precoz de las personas con factores de riesgo y un diagnóstico temprano de los afectados. Es importante la participación de la atención primaria de salud en la pesquisa de esta discapacitante enfermedad ocular.
ABSTRACT: The clinical, genetical and epidemiological characteristics of Retinitis Pigmentosa in families of Havana City.
We have made a descriptive, transversal investigation. The purpose is to know the results of the quest, classification, and multidisciplinary approach of retinitis pigmentosa in HavanaCity from 2006 to 2007.The individual files are revised as well as their charts and genealogic trees; all revisions were designed by The National Program of Retinitis Pigmentosa, to establish the clinical, genetical and epidemiological characteristics of Retinitis Pigmentosa in them. The perceptual resume is made out of the results showed in graphics.
Key Words: Retinitis Pigmentosa, Inheritance, Family, Nocturnal blindness
REFERENCIAS BIBLIOGRÁFICAS
1-Programa Nacional de Retinosis Pigmentaria MINSAP, 2004.
2-Gutiérrez Torres SM. Retinosis pigmentaria. Clasificación y Tratamiento. // retinosis.org /olo /libro rp / l. htm. [consultado julio 2007].
3-Kanski J. Oftalmología Clínica. Elsevier, Madrid. 2004.
4- Weleber RG, Gregory-Evans K. Retinitis pigmentosa and allied disorders. In: Ryan S.J. Editors. Elsevier Inc, Philadelphia. 2006.
5-Kaplan J, Bonneau D, Frézal J, MunnichA, Dufier JL. Clinical and Genetic Heterogeneity in Retinitis Pigmentosa. Human Genet. 1990; 85:635-642.
6- Herrera Mora M. Clasificación. En: Peláez Molina O. Retinosis Pigmentaria. Experiencia Cubana. Científico Técnica, La Habana.
7- Carpeta Metodológica de Atención Primaria de Salud y Medicina Familiar. Programa de Especialización Medicina General Integral. MINSAP. Ciencias Médicas, Ciudad
Habana. 1989.
8- Pérez Rojo N, Carmona Gutiérrez A. La familia y el nivel de salud de una comunidad. Aspectos conceptuales y Metodológicos. Rev. CubAdm. de Salud, 1982;8(3):23-27.
9- Pirech A. Epidemiology and prevalence of hereditary retinal Dystrophies in France. Jour France Ophthal. 1997;141(5):153-64.
10-Mäntyjärvi M, Tuppurainen K. Clinical Symptoms at Different Ages in Autosomal Dominant Retinitis Pigmentosa. Ophthalmologica. 1994;208:23-28.
11-Lisa M, Westin M, Carney Carol A.Genetic heterogeneity of Usher syndrome. Am J Hum Genet. 2000;6(Issue 6):1569-74.
12-Vaughan O. Oftalmología General. México DF, El Manual Moderno. 2002.
13- Cuba es ya un ejemplo de país en desarrollo con un Envejecimiento importante. En: http://www.sld.cu/sitios/gericuba. (Consultado mayo 2007).
Clasificación clínica de la Retinosis Pigmentaria:
Estadio I
Agudeza visual: (0.7 – 1.0) en el mejor ojo
Campo visual:mayor de 15° en el mejor ojo
Cristalino: Transparente
Catarata puntiforme subcapsular posterior
Vítreo: Flóculos oVítreo celular
Fondo de ojo: Disco óptico: Normal
Vasos: normales en calibre hasta el ecuador y discretamentedisminuidos en la periferia.
Mácula: Disminución o pérdida del reflejo foveal
Pigmentos: Escasos a predominio del sector nasal superior,ecuadory periferia. Aspecto gris verdoso de fondoo granular fino conservando coroides no visible de aparente aspecto normal.
Estadio II
Agudeza visual: (0.4 – 0.6) en el mejor ojo
Campo visual: Entre 11 – 15° en el mejor ojo
Cristalino: Transparente o Catarata subcapsular posterior parcial
Vítreo: Flóculos oVítreo fibrilar o desprendimiento de vítreo posterior.
Fondo de ojo: Disco óptico: Normal, ligeramente céreo
Vasos: Disminuidos de calibre en todo su trayecto.
Mácula: reflejo en baba de caracol, pliegues Peri maculares. Degeneración quística, zonas de atrofia epitelial con área central indemne.
Pigmentos: Abundantes en periferia y ecuador en todos los sectores.
Coroides: Daño ligero aspecto atigrado o no.
Estadio III.
Agudeza visual: (0.1 – 0.3) en el mejor ojo
Campo visual: Entre 10 y 5° en el mejor ojo
Cristalino: Transparente o Catarata subcapsular posterior subtotal
Vítreo: Múltiples flóculos, marcada degeneración, matriz posterior densa conopacidad blanquecina y fibras de interconexión. DVP
Fondo de ojo: Disco óptico: Palidez cérea
Vasos: marcada estrechez generalizada
Mácula: Atrofia del epitelio pigmentario central, edema quístico, agujero lamelar, pigmentos o distrofia o atrofia coriorretinianas
Pigmentos: Generalmente cubren todo el fondo, polo posterior, perivasculares y peripapilares.
Estadio IV
Agudeza visual: menor de 0.05 en el mejor ojo
Campo visual: menorde5° en el mejor ojo
Cristalino: Catarata subcapsular posterior subtotal, catarata total
Vítreo: múltiples flóculos, marcada degeneración fibrilar con desorganización de fibras que se mueven fácilmente, DVP, colapso de la matrizposterior opacificada, disminución del volumen del gel.
Fondo de ojo: Disco óptico: Subatrófico, atrófico.
Vasos: Marcadamente disminuidos de calibre con perivasculitishasta llegar a exangües
Mácula: Atrofia del epitelio pigmentario central,
Edema quístico, agujero macular, pigmentos, zonas de atrofiacoriorretiniana, degeneración severa
Pigmentos: Abundantes en ecuador, periferia, polo posterior, peripapilares en ocasiones
forman verdaderos enrejados y siguen el trayecto de los vasos coroideos.
Coroides con marcado daño como cordones oscuros y zonas de desnudez coroidea y observación de esclera.
CRITERIOS MAYORES: CAMPO VISUAL Y AGUDEZA VISUAL
-CLASIFICACIÓN DIAGNOSTICA DE LA ESCUELA CUBANA DE RETINOSIS PIGMENTARIA.
-SEGÚN LACLÍNICA: (FORMA LOCALIZACIÓN DE LOS PIGMENTOS)
TÍPICA
ATÍPICA: monocular, en sector, central o inversa, paravenosa o sin pigmentos.
ASOCIADA: acompañando a otras enfermedades como obesidad, sordera parcial o total, enfermedades neurológicas, etc.SON LOS SÍNDROMES.
-SEGÚN LA GENÉTICA: HERENCIA AUTOSÓMICA DOMINANTE
HERENCIA AUTOSÓMICA RECESIVA
HERENCIA RECESIVA LIGADA AL X
HERENCIA NO DEFINIDA
-SEGÚN EL ESTADIO DE LA EVOLUCIÓN DELA RP:
ESTADIO IESTADIO IIESTADIO IIIESTADIO IV
-SEGÚN LA EDAD DECOMIENZO DEL SÍNTOMA INICIAL:
DEBUT PRECOZ(antes de los 10 años)
DEBUT JUVENIL(entre 11 y 20 años)
DEBUT TARDÍO(a partir de los 21 años)
GRÁFICO 1. PATRÓN DE HERENCIA POR FAMILIAS CON RP
 |
GRÁFICO 2. RP SINDROMICA.CARACTERÍSTICAS CLÍNICAS Y EVOLUCIÓN.(51 PACIENTES)
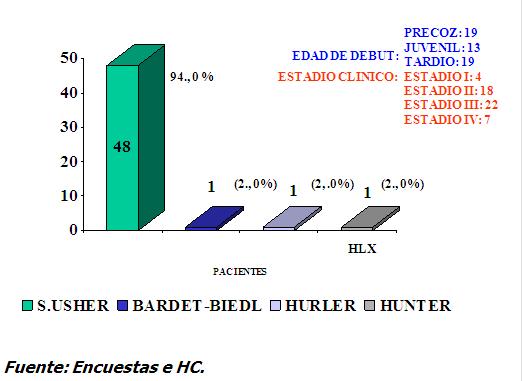 |
GRÁFICO 3. ESTADIOS CLÍNICOS POR FAMILIAS CON RP.
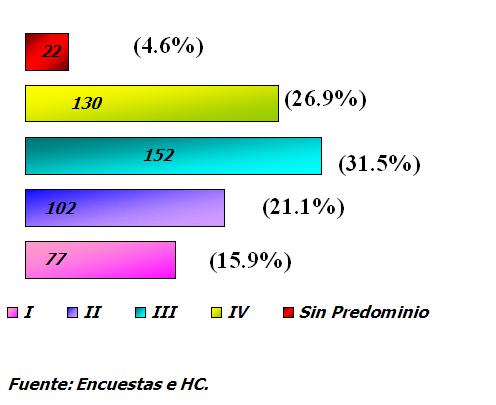 |
TABLA 1. ENFERMEDADES ASOCIADAS A LA RP POR PACIENTES
 |
PROGRAMA NACIONAL DE RETINOSIS PIGMENTARIA ENCUESTA INDIVIDUALFECHA:
No. HC ________No. de CI:_______________
NOMBRE:______________________________________ CÓDIGOFAMILIAR _______
DIRECCIÓN: ________________________________
LUGAR NAC: ______________
AÑO NAC: ________SEXO: ____
SÍNTOMA INICIAL: ____________
EDAD DE APARICIÓN:_______________
APP: Ninguno __ Miopía __ Glaucoma __ Catarata__ Retraso Mental __ Enanismo__
Hipoacusia __ D. mellitas __ HTA __ Sordomudez __ Polidactilia __Sindactilia _
Obesidad __Epilepsia__Trastornos del habla __ “Convulsiones” __ Asma __
Sarampión __ Varicela __ Parotiditis __ Rubéola __ Paludismo_____Otros _____
AFP:RP¿Quiénes? _____________________________
Miopía __ Consanguinidad___ Glaucoma __ Ciegos __ ¿Por qué?____________Mala Visión Nocturna_______
Epilepsia __ A. Bronquial __ D. Mellitas __ HTA __ Otras (especificar) _______
Hábitos Tóxicos: Fumador __Alcohol__Ser automedica___ Otras _________________
Examen Oftalmológico:
AV. (csc): ODOIPIO:OD: _____OI: _____
Nula ______
Bultos_______
PL______
Movimiento Manos____________
Cuenta dedos____________
Sentidocromático: Normal:_______Alterado: ______
| Refracción: | esf | cil | eje |
OD______ ________ _________
OI_______ ________ ________
GLAUCOMACampo visual Goldmann
__ Si __ No__Reducción concéntrica
__ Primario Ang. AbiertoODOI
__ Primario Ang. Estrecho< 5º_____
__ Secundario 5-10º_____
10-20°_____
CV de más de 20 grados:
ERG:Normal_____Subnormal_____Extinguido_____ODOI
Anexos:
Segmento anterior:
Medios:
CRISTALINO _____ VITREO
__ Transparente ___ Transparente
__ Cat. Sub. Post ___.Desp. V. Posterior
Fondo de ojo:
Disco:
Mácula:
Pigmentos:
Coroides:
Vasos retinianos:
HERENCIA: A. Dom____ A. Rec ____ Rec.Ligada X ____
No definida _____Simple____Múltiple_______
Desconocida_______
DIAGNÓSTICO: Retinosis Pigmentaria Típica____
Atípica (especificar tipo) _________________
Asociada (especificar tipo)________________
Estadio I __ II__ III__ IV___
Retinosis Pigmentaria Atípica_____
Retinosis Pigmentaria Asociada______
Debut: Precoz ____ Juvenil ____ Tardio ____

