










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Habanera de Ciencias Médicas
versión On-line ISSN 1729-519X
Rev haban cienc méd v.9 n.1 Ciudad de La Habana ene.-mar. 2010
CIENCIAS BÁSICAS BIOMÉDICAS
Universidad de Ciencias Médicas de La Habana (UCMH)
Centro Nacional de Genética Médica
Haplotipo del gen del factor VIII en el diagnostico molecular de la hemofilia A. Estudio de una familia
Factor VIII gene haplotype in the haemophilia a molecular dagnosis. A family study
Yaixa Piloto-Roque1,Arlet Acanda de la Rocha2, Ismael Cervera García3, Yulia Clark Feoktistova4, Teresa Collazo-Mesa5
Centro Colaborador de la OMS para el Desarrollo de Enfoques Genéticos en la Promoción de Salud. Calle 146 núm. 3102 esq. Ave 31. Marianao. Ciudad de La Habana. Cuba.
Laboratorio Biología Molecular. Teléfonos: 208 99 91/97 ext. 1042-1047.
1Lic. Bioquímica. Aspirante a Investigador. Instructora. yaixa.piloto@infomed.sld.cu
2Lic. Bioquímica. Aspirante a Investigador. arlet@cngen.sld.cu
3Lic. Laboratorio Clínico. Instructor. icervera@infomed.sld.cu
4Lic. Bioquímica. Aspirante a Investigador. yulia.clark@cngen.sld.cu
5Lic. Bioquímica. Investigadora Auxiliar. tcollazo@infomed.sld.cu
RESUMEN
La hemofilia A se caracteriza por ser una enfermedad congénita del trastorno de la coagulación y constituye un desorden recesivo ligado al cromosoma X. El estudio molecular se realiza por estudio indirecto, por ser causada por mutaciones heterogéneas en el gen del factor VIII. Se estudió una familia afectada, para la detección de portadora de la gestante y posteriormente se realizó el diagnóstico prenatal. La extracción de ADN se hizo por el método de precipitación salina Salting Out a tres muestras de sangre y una de líquido amniótico. Se efectuó el análisis de los polimorfismos St14, Bcl I y Hind III. Para la determinación de sexo fetal, se estudió el gen AMXY. La técnica empleada fue la reacción en cadena de la polimerasa.
El análisis del marcador Bcl I arrojó que la gestante era portadora de hemofilia A, pero al ser homocigótica no era informativa; el polimorfismo St14 por sí solo no brindaba la información de la condición de la gestante, pero al ser heterocigótica para el mismo y conociendo de antemano la información de ser portadora, se pudo realizar diagnóstico prenatal, gracias al análisis conjunto de los marcadores. El polimorfismo Hind III no fue informativo. El feto resultó ser varón sano.
Palabras clave: Hemofilia A, Bcl I, St14, Hind III, factor VIII.
ABSTRACT
Hemophilia A is a coagulation disorder congenital disease which consist in a recessive disorder linked to X chromosome. HA is cause by heterogeneous mutations in factor VIII gen, that´s why, the study was carry out by indirect studies. We studied one family afected; we were determinated of carrier status of pregnancy woman and later we relizated of prenatal diagnosis. The DNA extraction from the three blood samples and one amniotic fluid was obtained by the saline precipitation procedure (Salting Out).
We studied the polymorphisms St14, Hind III and Bcl1. The determination of fetal sex was studied of AMXY gen. The technique used was Polymerase Chain Reaction.
The Bcl I marker analysis showed that the mother was a carrier of haemophilia A but being homozygous was not informative, ST14 polymorphism alone did not provide information on the condition of the mother but to be heterozygous for it and knowing beforehand information of being a carrier prenatal diagnosis was possible thanks to joint analysis of the markers. Hind III polymorphism was not informative. The male fetus was found to be healthy.
Key words: Haemophilia A, Bcl I, St14, Hind III, factor VIII.
INTRODUCCIÓN
La hemofilia A (hemofilia clásica, HA), enfermedad recesiva ligada al cromosoma X, es la coagulopatía congénita severa más común; se debe a la deficiencia o ausencia del factor VIII (FVIII). Debido a su patrón de herencia, suele afectar casi exclusivamente a los varones, aunque existen reportes de mujeres heterocigóticas. Presenta una incidencia de 1:5 000 a 1: 10 000 varones.1
El gen de FVIII tiene 186 kb de longitud; es uno de los genes más largos caracterizados hasta el presente, contiene 26 exones (con un rango de longitudes que va desde 69 a 3106 pares de bases, pb) y 25 intrones (algunos tan largos como 32,4 kb) y está localizado en la región distal del brazo largo del cromosoma X a nivel de la banda Xq28. Este gen produce un RNAm de 9 kb.1,2
La HA no presenta ninguna alteración visible con métodos citogenéticos, y además el análisis a nivel molecular ha sido muy difícil debido al gran tamaño del gen, a su compleja organización genómica y a la heterogeneidad alélica detectada en los distintos individuos. En la actualidad, se han reportado 65 % de mutaciones puntuales, 28 % de deleciones y 6 % de inserciones.1,2
La detección de portadoras se pudiera abordar por métodos directos e indirectos. El diagnóstico directo caracteriza la alteración molecular en los pacientes y sus familias. Sin embargo, este análisis es muy complicado en esta enfermedad, pues este gen es uno de los más grandes del genoma y la mayoría de las mutaciones son puntuales y la frecuencia de las mismas es menor de 1 %; excepto una mutación compleja que consiste en una inversión a nivel del intrón 22 que, según reporta la literatura, afecta aproximadamente a 50 % de los enfermos.3
Por esta razón, el estudio indirecto usando marcadores polimórficos asociados al gen del factor VIII, es la mejor opción.
En Cuba, para la detección de portadoras y diagnóstico prenatal de la HA se emplean los marcadores moleculares intragénicos Bcl I y Hind III; ambos son polimorfismos de longitud de fragmentos de restricción (RFLP`s) y se localizan en el intrón 18 y 19, respectivamente. El otro polimorfismo usado es St14, el cual se encuentra en el locus DXS 52, es un VNTR (repeticiones en tándem de número variable), el cual presenta hasta 14 alelos, según reporta la literatura.4-6
MATERIAL Y MÉTODOS
Se estudió una familia diagnosticada clínica y bioquímicamente. El universo de estudio estuvo conformado por la gestante que es el propósito del estudio, el hermano hemofílico, o sea, el enfermo y la madre. No fue posible estudiar la muestra del padre. Se realizó la extracción de ADN por el método de precipitación salina Salting Out 7 a partir de 10 mL de sangre periférica con EDTA 56 mg/mL como anticoagulante y 20 mL del líquido amniótico.
Se usó la técnica de reacción en cadena de la polimerasa (RCP) y posterior digestión enzimática en los polimorfismos de longitud de fragmentos de restricción Bcl I y Hind III; se visualizaron los resultados en gel de agarosa a 3 %. El producto de amplificación del polimorfismo St14 se testó directamente en gel de agarosa a 2 %. Las condiciones de PCR y digestiones enzimáticas se realizaron según lo descrito por Scott y col.5 La determinación del sexo fetal se hizo estudiando el gen AM-XY, técnica descrita por Mehra y col.8
RESULTADOS
En la figura 1, se muestra el árbol genealógico de la familia, analizada junto con el haplotipo de los marcadores moleculares analizados. Para Bcl 1, el enfermo tiene el alelo 2 del marcador y la gestante es homocigótica para el mismo.(figura 2). Para los polimorfismos St14 y Hind III, el enfermo posee el alelo 1 para cada marcador respectivamente; sin embargo, para St14 la embarazada es heterocigótica (1/3) y para Hind III es homocigótica (1/1).

Figura 1. Árbol genealógico de la familia analizada. Al lado de cada miembro estudiado: I.1, II.1, II.2 se muestran los alelos encontrados de los marcadores Bcl I, St14 y Hind III, respectivamente.
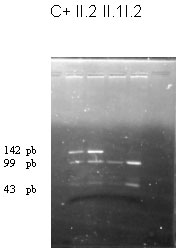
Figura 2. Gel de agarosa a 3 %, mostrando el polimorfismo Bcl I. Carrilera 1 (c+): control positivo heterocigótico, la enzima Bcl I y se obtienen las bandas correspondientes a 142, 99 y 43 pb. Carrilera II. 2: Madre de la gestante, presenta las 3 bandas, heterocigótica. Carrilera II.1: hemofílico, alelo 2 (bandas 99 y 43 pb). Carrilera I.2. Gestante homocigótica para el alelo 2 (bandas 99 y 43 pb).
Al efectuar la determinación de sexo resultó ser un feto varón. Las bandas halladas en el estudio del gen AMXY fueron de 1 000 pb, 800 pb y 470 pb, tal como se muestra en la figura 3.

A: control hembra normal; B: caso en estudio; C: control varón normal
Las bandas observadas corresponden a 1 000 pb, alelo en el cromosoma X; 800 pb, alelo en el cromosoma Y; 470 pb, control interno de amplificación. (Es indudable que en nuestro paciente las bandas son las esperadas en un varón).
Cuando se realizó el diagnóstico prenatal, se estudió el marcador St14; el feto presentó el alelo 3 para este marcador.(figura 4).
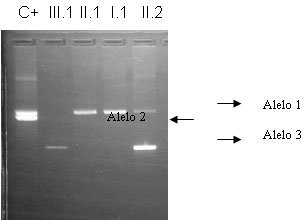
Figura 4. Gel de agarosa al 2 % mostrando el polimorfismo St14. Carrilera 1: control positivo. Carrilera 2 (III.1): feto varón sano (alelo 3). Carrilera 3 (II.1): hemofílico, presenta el alelo 1, Carrilera 4 (I.1): madre de la gestante, homocigótica para el alelo 1, Carrilera 5 (II.2): Gestante, presenta el alelo 1 y 3.
DISCUSIÓN
Al analizar al polimorfismo Bcl I, se establece que el gen afectado se está segregando junto con el alelo 2 de dicho marcador, como se muestra en la Figura 2. Para la realización de los estudios indirectos en general, es necesario analizar varios miembros de la familia afectada; es imprescindible el enfermo para establecer la fase de ligamiento, pero además los padres y en caso de la hemofilia, debido a su patrón de herencia, en ocasiones es imprescindible estudiar hasta los abuelos. La obtención de muestras de los miembros de la familia como de la paciente se realizan bajo las normas éticas. No fue posible estudiar al padre de la gestante, pero ella resultó ser 2/2; por tanto, se infiere que heredó el alelo 2 del padre y el 2 de la madre, lo cual determina su condición de portadora del alelo afectado, pero al ser homocigótica no es informativa. Con el estudio del polimorfismo
St 14, no se puede conocer si la gestante es portadora o no, debido a que su madre es homocigótica, pero, sin embargo, se conoce que es portadora por la información que ofreció el otro marcador y la gestante es heterocigótica, por tanto, se puede realizar diagnóstico prenatal.
El polimorfismo Hind III fue no informativo; como se observa en la Figura 1, la gestante y la madre son homocigóticas.
El diagnóstico prenatal se realizó estudiando al marcador St14, pues era el único para el cual la gestante era heterocigótica; el feto presentó el alelo 3 para este marcador (Figura 4); por lo tanto, es un feto varón sano.
Con el estudio de estos tres marcadores, se resalta la importancia de estudiar varios polimorfismos para la detección de portadoras y el diagnóstico prenatal de la hemofilia A. El estudio directo nos permite detectar la mutación en sí que está provocando la enfermedad, pero esta entidad es de gran heterogeneidad alélica y además el porciento de las mutaciones es menor de 1 %; es imposible determinar cada una de las mutaciones reportadas en el gen afectado y, por otra parte, la secuencia es una técnica muy cara. La única mutación candidata a estudiar es la inversión del intrón 22, que según reporta la literatura su frecuencia es de 50 % en pacientes con hemofilia A severa. 9,10 Con el análisis de esta familia, presenciamos cómo la información de los polimorfismos se complementan entre sí. En enfermedades como la hemofilia es muy importante el estudio por métodos indirectos, pues nos posibilita llegar a un diagnóstico en un corto período de tiempo, ya que el análisis a nivel molecular es muy difícil debido al gran tamaño del gen y su compleja organización genómica. De ahí, la sugerencia de estudiar otros polimorfismos del gen del factor VIII para garantizar un mayor porciento de diagnóstico y con esto elevar la calidad del estudio molecular de las familias afectadas, tales como: VNTR del intrón 13 y del intrón 22.11-14
CONCLUSIONES
Con el estudio de los marcadores moleculares Bcl I, Hind III y St14, se determinó la condición de portadora de hemofilia A de la gestante. Además, fue informativa para el marcador St14 y, por tanto, se realizó el diagnóstico prenatal. Resultó ser un feto varón sano.
REFERENCIAS BIBLIOGRÁFICAS
1. Wood WI, Capon DJ, Simonsen CC. et al. Expression of active human factor VIII from recombinant DNA clones. Nature. (312):330-6;1984.
2. Vehar GA, Keyt B, Eaton D. et al. Structure of human factor VIII. Nature. (312):337-42;1984.
3. Liu Q, Sommer SS. Subcycling PCR for multiplex long-distance amplification of regions with high and low GC content: application to the inversion hotstop in the factor VIII gene. Biot. (25): 1022-28;1998.
4. Oberle IG, Camerino R, Heilig L, Grunebaum JP,Cazenave C, Crapanzano PM, Mannucci & JLMandel. Genetic screening for haemohilia A with a polymorphic DNA probe. N. Engl. J. Med. (312): 682-686;1985.
5. Scott C. Kogan and Jane Gitschier. Genetic prediction of Hemophilia A. In: Michael A. Innis, David H. Gelfand, John J. Sninsky, Thomas J White. PCR Protocols. A guide to methods and applications; 1990, p. 288-299.
6. Xuefeng W, Yuanfang L, Zhiguang L, Haiyan C, Xiaujie S, Yishi F. et al. Carrier detection and prenatal diagnosis of hemophilia A. Clin Chem Lab Med. (39): 1204-1208;2001.
7. Miller, SA. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 16 (3): 1215;1988.
8. Mehra S, Schmid W, Spiegel R. Rapid detection of sex chromosomal aneuploidies by PCR. Indian J Med Res.(101):111-4;1995, Mar.
9. Lombardi Am, Cabrio L, Zanon E, Pellati A, Vettore S, Ginolani A. Confirmation of the Value of a Modified Long-distance Polymerase Chain Reaction in the Detection of Inversion Intron 22 in Severe Hemophilia A: A Technical Note. Clin Appl Throtnbosis/Hemostasis. 11 (4):493-96;2005.
10. Ahmed R, Kannan M, Choudhry V, Saxena I. Mutation reports: Intron 1 and 22 inversions in Indian haemophilics. Ann Hematol. (82):546-547;2006.
11. Martínez GR, Benítez AH, Navarrete C, Peñaloza ER, Salamanca GF, Aranda AD. Polymorphism Distribution of Int13, Int22, and St14 VNTRs in a Mexican Population and Their Application in Carrier Diagnosis of Hemophilia A. Am Journ of Hematol. (77):1-6;2004.
12. Chodwdhyry MR, Twari M, Kabra M. Prenatal diagnosis in Hemophilia A using factor VIII gene polymorphism Indian experience. Ann Hematol. (82):427-43;2003.
13. Stonebraker JS, Brooker M, Amand R, Farrugias A, Srivastava A. A study of reported factor VIII use around the world. Haemophilia. (16):33-46;2010.
14. Jayandharan G, Nair S, Poonnoose M, Thomas R, John J, Keshov S. et al. Polymorphism in factor VII gene modifies phenotype of severe haemophilia. Haemophilia.(15):1228-1236;2009.

