










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Habanera de Ciencias Médicas
versión On-line ISSN 1729-519X
Rev haban cienc méd v.10 n.2 Ciudad de La Habana abr.-jun. 2011
CIENCIAS CLÍNICAS Y PATOLÓGICAS
Universidad de Ciencias Médicas de La Habana
Instituto de Ciencias Básicas y Preclínicas «Victoria de Girón»
El tumor de wilms. Un paradigma de heterogeneidad genética
Wilm's toumor: a paradigm of genetic heterogeneity
Rolando A. Hernández Fernández
Profesor Titular de Bioquímica. Especialista Segundo Grado en Bioquímica Clínica. Calle 208 núm. 1921 entre 19 y 21-A. Reparto Atabey. Municipio Playa. Ciudad de La Habana.Teléfono:271-7741. rolher@infomed.sld.cu
RESUMEN
El Tumor de Wilms constituye el más frecuente de los cánceres renales pediátricos, aparece antes de los 5 años de edad y con igual frecuencia en ambos sexos. El gen causante de la enfermedad está localizado en 11p13, se extiende unas 50 kb con 10 exones y sus alteraciones pueden ser tanto genéticas como epigenéticas. Por diferentes mecanismos se originan al menos 24 productos con funciones diversas. Existen otras regiones cromosómicas, cuyas alteraciones pueden dar lugar a la aparición del tumor. En este sentido, el Tumor de Wilms es un ejemplo sobresaliente de heterogeneidad genética. El presente trabajo hace un análisis de los fundamentos moleculares de la enfermedad y presenta a modo de ilustración una breve reseña de los principales síndromes hereditarios con predisposición a presentar este tumor.
Palabras clave: Tumor de Wilms, Heterogeneidad genética, Impronta genómica, Síndrome Denys-Drash, Síndrome WAGR, Síndrome Beckwith-Wiedeman, Síndrome Simpson-Golabi-Behmel, Síndrome Frasier.
ABSTRACT
Wilm's tumor is the most frequent cancer of the kidney in childhood with onset before five year old. WT gene was mapped in 11p13 and span 50 kb with 10 exons. WT modifications would be either genetics or epigenetics. The gene code for at least 24 isoforms of protein products. Other chromosomes loci whose alterations may be cause of disease have been identified, so Wilm's tumor is a remarkable paradigm of genetic heterogeneity. In this paper an analysis of the molecular basis of the disease is presented and, also, a brief references on main hereditary syndromes which include Wilm's tumor.
Key words: Wilm's tumor, genetic heterogeneity, genomic imprinting, Denys-Drash syndrome, WAGR syndrome, Beckwith-Wiedeman syndrome, Simpson-Golabi-Behmel syndrome, Frasier syndrome.
INTRODUCCIÓN
El Tumor de Wilms o nefroblastoma es el más frecuente de los tumores sólidos intrabdominales de la infancia y aparece en 1 de cada 10 000 niños. La edad media de diagnóstico es de 3 a 4 años con la mayoría de los niños ya afectados a la edad de 6 años. Aún cuando la mayoría de los tumores son esporádicos y unilaterales, los bilaterales y multifocales (varios en el mismo riñón) dan cuenta aproximadamente de 10% de los pacientes. Estos niños son diagnosticados antes que los que tienen tumores unilaterales.1
Se origina a partir de restos de riñones inmaduros, pues se detecta la persistencia de estructuras llamadas restos nefrogénicos, que no son visibles en los riñones normales después de un año, pero se pueden observar en 30% de los niños con tumores esporádicos y prácticamente en 100% de los tumores familiares. Para un estudio más detallado de la relación entre el Tumor de Wilms y el desarrollo del riñón ver el reciente trabajo de Rivera y Haber.2 La forma familiar del Tumor de Wilms como entidad única es rara. Solamente, 2% tienen un familiar afectado y la predisposición al tumor se segrega como un rasgo autosómico dominante. 3 Sin embargo, existen varios síndromes asociados con predisposición a este tumor. Entre ellos los síndromes WAGR (por Tumor de Wilms, Aniridia, anomalías Génito-urinarias y Retraso mental), Denys-Drash (DDS), Beckwith-Wiedeman (BWS), Frasier, Simpson-Golabi-Behmel (SGBS) y el Tumor de Wilms familiar. En el presente trabajo, se exponen los fundamentos moleculares de la enfermedad y algunos síndromes asociados haciendo énfasis, en los casos posibles, en la relación genotipo fenotipo.
DESARROLLO
Presentamos una descripción de los aspectos moleculares relacionados con la génesis del Tumor de Wilms.
Genética. El Tumor de Wilms es un ejemplo de heterogeneidad genética, pues mutaciones en varios genes pueden dar lugar a la enfermedad. También es un ejemplo de cómo a partir de la información contenida en un gen pueden formarse varias proteínas incluso con funciones diferentes. Por su forma de presentación, se corresponde con el modelo de los dos eventos desarrollado por Knudson y, por lo tanto, WT1 (denominado así por el ser el primer gen vinculado a la génesis del Tumor de Wilms) se trata de un gen supresor tumoral.
La localización del gen se produjo a partir de los casos con aniridia, que está presente en 1 % de los pacientes con Tumor de Wilms en comparación con la frecuencia de 1 en 50 000 en la población general. Los estudios citogenéticos en la mayoría de los pacientes con WAGR tenían un borramiento visible en la banda 11p13 y esto permitió identificar en esa zona del gen causante de la aniridia, PAX6, y del Tumor de Wilms (WT1). A este hallazgo también contribuyó el hecho de que el Tumor de Wilms esporádico se asociaba a la pérdida de heterocigocidad en 11p. El gen fue asilado posteriormente por varios grupos mediante clonación posicional en 1990.
El gen WT1 se extiende unas 50 kb en la región 11p13 y tiene 10 exones y se expresa en varias proteínas debido a un empalme alternativo, por mecanismos de edición del ARNm y por la utilización de codones de iniciación alternativos durante la traducción. El promotor de WT1 carece del motivo TATA y es rico en GC y es autorreprimido por WT1 en ensayos de transfección. Una descripción detallada de las características del gen y de sus principales productos ha sido publicada recientemente.4
En los tumores esporádicos aparecen mutaciones inactivantes en 10-15 % de WT1 mientras que en 10% se producen mutaciones activantes del gen de la â-catenina,5 que juntos constituyen aproximadamente 15%, lo cual implica la participación de la vía de transducción de señales de WNT en la patogénesis del tumor.6 Wnt-4 es una glicoproteína de secreción que funciona como un autoinductor en la transición del mesénquima al epitelio y en el desarrollo normal de la neurona. También en estos casos, una tercera parte muestran pérdida de la impronta del IGF2 de lo que se deduce que esta alteración es importante en el desarrollo del tumor.
Teniendo en cuenta que solamente en 20 % de los casos se han encontrado mutaciones en WT1 se ha investigado en busca de otros genes cuyas mutaciones pudieran contribuir a la aparición de esta neoplasia.
Durante la década siguiente a la identificación de WT1, otros genes fueron descritos relacionados con la génesis de la enfermedad. Se ha reportado pérdida de heterocigocidad en 11p en 40-50% de los casos (gen WT2); en 16q (WT3) en 20 %; en 1p y en 11q en 30 %. En 11p15, hay genes con impronta implicados en la génesis del tumor entre ellos el causante de BWS. Modificaciones en 7p15-p11.2 (WT5) aparecen en 10%. Todos estos hallazgos están descritos en el excelente trabajo de Brown y Malik. 7 Más recientemente, se han localizado otros sitios relacionados con el tumor en Xq11.1 (WTX) que aparece mutado con una frecuencia de 7 % a 29 %, según la población estudiada 8 y en 11q13.4 (SKCG-1 por Sporadic Kidney Cander Gene). 9 El tipo familiar ha sido asociado a cambios en 17q12q21 (FWT1 o WT4) y en 19q13.4 (FWT2).10
Además, se han encontrado alteraciones en los mecanismos de impronta tanto en las regiones 11p13 y 11p15.5 11 como en 14q32. 12 La modificación de 11p casi siempre implica la pérdida del alelo materno. La región 11p15 contiene el gen del factor de crecimiento IGF2 (expresión paterna) y los genes inhibidores del crecimiento H19 y CDKNIC (expresión materna). De esta forma, la pérdida de impronta determina la expresión bialélica de IGF2 y la supresión de la expresión de H19 y CDKNIC 13,14
Otras alteraciones incluyen a los genes TP53 (p53) y CTNNB1 (â-CATENINA) en estados avanzados del tumor 15 e interacciones con el poderoso encogen c-Myc. 16
En humanos, el borramiento de un alelo de WT1 causa las anormalidades genitourinarias en el síndrome WAGR (con un riesgo de 30% del tumor), mientras que mutaciones con cambio de lectura en la línea germinal de heterocigóticos conduce a los defectos más severos observados en DDS (con 90% del riesgo del tumor), probablemente, debido a un efecto dominante negativo de la proteína mutada. En estos tumores WT1 parece actuar como el clásico gen supresor tumoral con pérdida de función. La mutación de WT1 representa un evento temprano porque sus mutaciones han sido encontradas en restos nefrogénicos, los cuales son precursores premalignos de WT.
Aún cuando las alteraciones de WT1 son muy frecuentes en el Tumor de Wilms y por ello se les considera como la causa principal; también sus modificaciones pueden aparecer en otros tipos de neoplasias. Por métodos inmunohistoquímicos se ha podido detectar la presencia de pWT tanto en tumores sólidos como en neoplasias del sistema hemolinfopoiético, especialmente en leucemias.17
La proteína WT1 (pWT1).- El gen WT1 codifica varias isoformas de una proteína de 52 ó 56 kDa (pWT1). La más corta se produce por empalme alternativo que excluye al exon 5 que codifica un segmento de 17 aminoácidos en el dominio amino terminal. No obstante todas las isoformas presentan una región aminoterminal rica en glutamina y prolina, posiblemente con función de transactivación, y una región carboxilo terminal de unión al ADN o ARN que contiene cuatro estructuras digitiformes de Zn2+, cada una codificada por los exones del 7 al 10, que son similares a los encontrados en la familia de los factores de transcripción EGR (del inglés early growth response).18 Existe otro empalme alternativo que determina la inclusión o exclusión del tripéptido KTS [lisina (K), treonina (T) y serina (S)] entre la tercera y cuarta estructuras de Zn2+. 7 Esto trae como resultado la formación de al menos cuatro productos génicos: (E5+)-(KTS+); (E5+)-(KTS-); (E5-)-(KTS+) y (E5-)-(KTS-). Aunque aún no se conoce totalmente la importancia funcional de estas isoformas estudios recientes han demostrado que la isoforma que contiene el exon 5 (E5+) produce una resistencia a la apoptosis en algunos tipos de leucemia 19 y que la isoforma que carece de KTS (KTS-) modula la expresión del gen de la renina. 20 La presencia o no de KTS parece ser lo más importante para la función general pues la forma KTS- funciona como un factor de transcripción, mientras que la KTS+ está implicada en el procesamiento de los ARNm posiblemente porque la inclusión de KTS produce una separación mayor entre las estructuras de Zn2`+ que impide la correcta unión al ADN. La pWT es modificada postraduccionalmente por fosforilación, ubiquitinación y sumoilación, 21,22 pero el significado de estas modificaciones se desconoce. En la Figura 1 se muestra un esquema resumen del gen WT1 y sus productos.
La proteína pWT1 puede unirse al ADN en regiones ricas en GC (5´GCGTGGGAGT-3´) conocidas como WTE (WT response element), (Figura 1) así como a largas secuencias ricas en CT. Aunque se han identificado muchos genes como blancos de la acción de pWT1, entre ellos el gen que codifica la anfiregulina y Bcl-2 solo existen evidencia de un papel directo de WT1 en la inducción de los genes Snail y Cdh1. 23 La actividad de la pWT1 es modulada por interacciones entre proteínas, entre ellas la proteína p53.
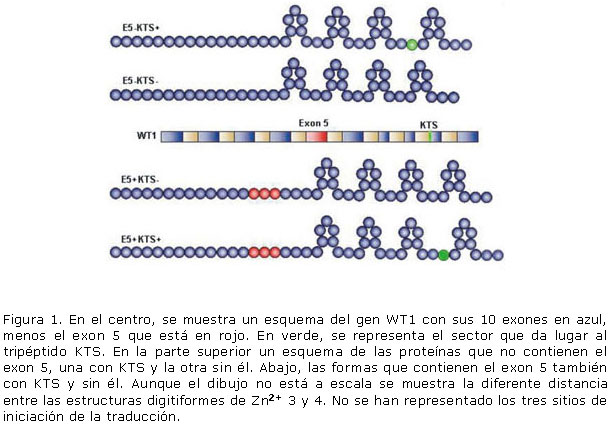
Mecanismo de acción de WT.- Como factor de transcripción WT funciona estimulando la expresión de genes específicos que están relacionados con la formación del riñón. WT1 es expresado normalmente durante el desarrollo del sistema génito-urinario. Sus niveles de expresión son bajos en el mesénquima metanéfrico inducido, se incrementan en la medida que las células experimentan diferenciación epitelial y se van atenuando en la misma medida que ellas maduran.24
Los datos presentados anteriormente indican la existencia de múltiples loci en la patogénesis del Tumor de Wilms. Sin embargo, la aparición tan temprana del tumor muestra que es muy probable que exista un número muy reducido de pasos limitantes, al contrario de lo que ocurre en los cánceres de los adultos. Hasta ahora la única correlación cierta entre genotipo y fenotipo es la existencia de mutaciones en WT1 en tumores con predominio del estroma, mientras que las mutaciones de p53 se observan en tumores con anaplasia. Los estudios de secuencia muestran que los cambios genéticos o epigenéticos en 11p se han encontrado en los restos nefrogénicos, donde no se ha hallado pérdida de heterocigocidad en 16p; lo cual implica que los eventos en 11p son anteriores a los de 16p en el desarrollo del tumor y producen un fenotipo más agresivo. Los productos de los genes implicados en la patogénesis del Tumor de Wilms pudieran estar relacionados con procesos diferentes (proliferación, diferenciación, apoptosis), pero pudieran estar interrelacionados, por ejemplo, ser genes controlados por WT1 o ser controladores de este gen, o genes que codifican proteínas que interactúan con WT.
En una versión simplificada de la patogénesis del Tumor de Wilms se pudiera plantear que al estimular la expresión de genes cuyos productos estimulan la proliferación celular tales como IGF-2 o disminuir la de genes que la inhiben como CDKNIC favorecen la proliferación incontrolada. Si por otra parte se estimula la expresión de Bcl-2 que tiene una función antiapoptótica, esto explica la proliferación de células en estados intermedios de diferenciación que normalmente deben ser eliminadas mediante la apoptosis. La desregulación de la â-catenina y la inactivación de p53 se sumarían a este fondo genético para el desarrollo del tumor.
El Tumor de Wilms sindrómico.- Además de su forma de presentación como entidad única, familial o esporádica, esta neoplasia se presenta formando parte de algunos síndromes genéticos. Estos síndromes se presentan sintéticamente a continuación establecidos en orden de frecuencia descendente de aparición del tumor.
El Síndrome Denys-Drash.- Los pacientes con este síndrome presentan trastornos genitales (pseudohermafroditismo, gonadoblastoma, hipertensión y enfermedad degenerativa renal) y 90% de riesgo de aparición de Tumor de Wilms. Recientemente, se ha vinculado la mutación R366H con la aparición de hernia diafragmática congénita. 25 Se ha encontrado pérdida de heterocigocidad en la zona 11p como en otros tipos de Tumor de Wilms. Se ha postulado la existencia de mutaciones dominantes negativas del gen WT1 como causantes del síndrome Denys-Drash.26
El Síndrome WAGR.- Esta entidad se caracteriza por un riesgo de aparición de Tumor de Wilms de 30%. Se debe a un micro-borramiento del locus 11p13 donde además del gen WT1 está ubicado el PAX6, cuyo producto tiene un papel fundamental en el desarrollo ocular y el nervioso, 27 Estudios posteriores mostraron que son las mutaciones del WT1 las causantes de las anormalidades génito-urinarias, pues este gen se expresa tanto en el desarrollo del riñón como de las gónadas. También han sido descritas otras anormalidades oculares y genitales.28 Puede estar acompañado de obesidad y en estos casos se ha sugerido usar las siglas WAGRO.29
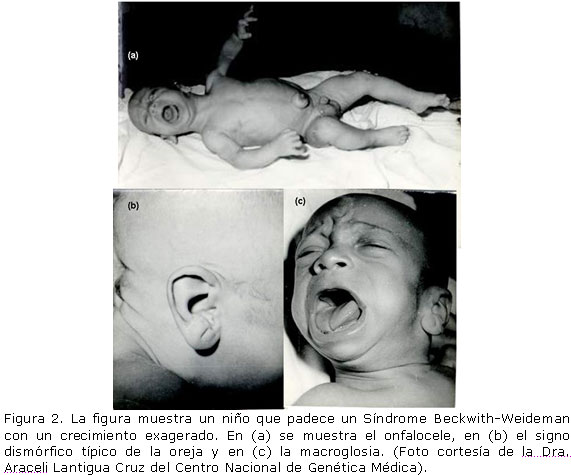
El Síndrome Beckwith-Wiedeman.- Se trata de un síndrome de hipercrecimiento con Tumor de Wilms en 5% de los casos.
Se origina como consecuencia de una disomía uniparental de una porción del cromosoma 11 (11p15.5) donde se localiza el gen de IGF2 que es un factor de crecimiento fetal y está sobrexpresado en el Tumor de Wilms. 13,14 Los casos familiares han permitido identificar la región 11p15 y algunos casos esporádicos tienen anormalidades cromosómicas en esa misma región. En los casos hereditarios, la transmisión es preferentemente materna y en los casos esporádicos predomina la disomía uniparental paterna, duplicaciones cromosómicas paternas y translocaciones maternas. Todos estos datos implican defectos de impronta en BWS. 30 Además la pérdida constitucional de la impronta del IGF2 ha sido encontrada en varios pacientes con BWS. Las lesiones moleculares precisas que causan BWS son complejas e implican dos regiones con metilación diferencial (DMR) en 11p15, centradas alrededor de H19, y de CDKNIC que son inhibidores del crecimiento. Para más información ver el reciente trabajo de Weksberg, Shuman y Beckwith. 31 En la Figura 2, se muestran algunas características de este síndrome.
El Síndrome Simpson-Golabi-Behmel.- Se trata también de un síndrome de hipercrecimiento pero ligado al cromsoma X. Muestra similitudes fenotípicas con el síndrome Beckwith-Weidemann y con otros caracterizados por un crecimiento excesivo.32 (Figura 3). El sobrecrecimiento es prenatal con peso al nacer de 5 kg o más, estatura superior a los 50 cm pero con el tiempo la edad ósea se normaliza. 33 El gen cuyas mutaciones son causa de la enfermedad fue localizado en la región Xq26, donde se localizan los genes de las glipicanas 3 y 4 (GPC3 y GPC4). Se han identificado mutaciones somáticas 34 y mutilaciones 35 del gen GPC3, así comomosaicismo germinal 36 en familias con syndrome Simpson-Golabi-Behmel.

El Síndrome Frasier.- Lo más característico del síndrome es la disgenesia gonadal y la insuficiencia renal crónica. Se han identificado mutaciones en el sitio donador para el empalme alternativo del intrón 9 del gen WT1 37 lo cual produce una relación alterada entre las dos formas de empalme (KTS+ y KTS-) de pWT1. El riesgo de Tumor de Wilms en el síndrome Frasier es menor que en el Denys-Drash.
Sobre el tratamiento.- Los nuevos protocolos de tratamiento dan buenos resultados en 85% de los casos, pero aún existe 15% que muere por causa de la enfermedad. Nuevos procedimientos terapéuticos son necesarios para estos niños y para una intervención más fácil y segura de todos los enfermos. 38 No es posible descartar el futuro uso de la terapia génica y el diagnóstico preimplantación acompañado del consejo genético a la familia.
CONCLUSIONES
El Tumor de Wilms constituye un ejemplo elocuente de los mecanismos de heterogeneidad genética tanto en la génesis del cáncer como de otros trastornos. La sola identificación de varios loci genéticos implicados en el origen de la enfermedad es más que suficiente para confirmar esta idea. Pero solamente el análisis detallado del WT1 nos ofrece los argumentos necesarios para confirmar la heterogeneidad en el fundamento molecular del Tumor de Wilms. Un solo gen de 50 kb es capaz de codificar al menos 24 productos diferentes por mecanismo de empalme alternativo, edición del ARNm y por el uso de diferentes sitios de inicio de la traducción. El hecho de que las formas principales (con o sin exon 5 y con o sin RTS) se produzcan en una proporción más o menos constante, indica la existencia de finos mecanismos de regulación de la expresión del gen. Los conocimientos acerca de los mecanismos moleculares implicados en la génesis del Tumor de Wilms deben conducir en los próximos al diseño de protocolos terapéuticos más eficaces y más útiles en el tratamiento de las numerosas formas de presentación de esta enfermedad.
REFERENCIAS BIBLIOGRÁFICAS
1. Stiller, CA. Epidemiology and genetics of childhood cancer. Oncogene. 2004; 23: 64296444.
2. Rivera, MN y Haber, DA. Wilms' Tumour: Connecting tumorigenesis and organ development in the kidney. Nature Rev Cancer. 2005; 5: 699-712.
3. Breslow NE, Beckwith JB, Perlman EJ, y Reeve AE. Age distributions, birth weights, nephrogenic rests, and heterogeneity in the pathogenesis of Wilms tumor. Pediatr. Blood Cancer. 2006; 47, 260267.
4. Yang L, Han Y, Suárez Saíz F. y Minden MD. A tumor suppressor and oncogene: the WT1 story. Leukemia. 2007; 21: 868876.
5. Royer-Pokora B, Weirich A, Schumacher V, Uschkereit C, Beier M, Leuschner I, et al. Clinical relevance of mutations in the Wilms tumor suppressor 1 gene WT1 and the cadherin-associated protein â1 gene CTNNB1 for patients with Wilms tumors: results of long-term surveillance of 71 patients from International Society of Pediatric Oncology Study 9/Society for Pediatric Oncology. Cancer. 2008; 113, 10801089.
6. Fukuzawa R, Anaka MR, Weeks RJ, Morison IM. y Reeve A E. Canonical WNT signalling determines lineage specificity in Wilms tumour. Oncogene. 2009; 28: 10631075.
7. Brown KW y Malik KTA. The molecular biology of Wilms´tumour. Cambridge University Press. 14 May 2001 Disponible en: http://www-ermm.cbcu.cam.ac.uk
8. Wegert J, Wittmann S, Leuschner I, Geissinger E, Graf N. y Gessler. WTX inactivation is a frequent, but late event in Wilms tumors without apparent clinical impact. Genes Chromosomes Cancer. 2009; 48, 1102-1111.
9. Singh KP y Roy D. SKCG-1: a new candidate growth regulatory gene at chromosome 11q23.2 in human sporadic Wilms tumours. Br J Cancer. 2006; 94: 1524-1532.
10. Rapley EA, Barfoot R, Bonaïti-Pellié C, Chompret A, Foulkes W, Perusinghe N. et al. Evidence for susceptibility genes to familial Wilms tumour in addition to WT1, FWT1 and FWT2. Br J Cancer. 2000; 83(2): 177183.
11. Satoh Y, Nakadate H, Nakagawachi T, Higashimoto K, Joh K, Masaki Z. et al. Genetic and epigenetic alterations on the short arm of chromosome 11 are involved in a majority of sporadic Wilms' tumours. Br J Cancer. 2006; 95: 541 547.
12. Astuti D, Latif F, Wagner F, Gentle D, Cooper WN, Catchpoole D. et al. Epigenetic alteration at the DLK1-GTL2 imprinted domain in human neoplasia: analysis of neuroblastoma, phaeochromocytoma and Wilms' tumour. Br J Cancer. 2005; 92: 1574-1580.
13. Sparago A, Cerrato F, Vernucci M, Ferrero GB, Silengo MC. y Riccio A. Microdeletions in the human H19 DMR result in loss of IGF2 imprinting and Beckwith-Wiedemann syndrome. Nature Genet. 2004; 36, 958-960.
14. Prawitt D, Enklaar T, Gärtner-Rupprecht B, Spangenberg C, Oswald M, Lausch E, et al. Microdeletion of target sites for insulator protein CTCF in a chromosome 11p15 imprinting center in Beckwith-Wiedemann syndrome and Wilms' tumor. Proc. Natl Acad. Sci. USA. 2005; 102, 4085-4090.
15. Ruteshouser EC, Robinson SM. y Huff V. Wilms tumor genetics: mutations in WT1, WTX, and CTNNB1 account for only about one-third of tumors. Genes Chromosomes Cancer. 2008; 47, 461-470.
16. Han Y, San-Marina S, Liu J. y Minden MD. Transcriptional activation of c-myc proto-oncogene by WT1 protein. Oncogene. 2004; 23: 69336941.
17. Nakatsuka S, Oji Y, Horiuchi T, Kanda T, Kitagawa M, Takeuchi T, et al. Immunohistochemical detection of WT1 protein in a variety of cancer cells. Modern Pathol. 2006; 19: 804-814.
18. Stoll R, Lee BM, Debler EW, Laity JH, Wilson IA, Dyson HJ. y Wright, PE. Structure of the Wilms tumor suppressor protein zinc finger domain bound to DNA. J. Mol. Biol.2007; 372, 1227-1245.
19. Ito K, Oji Y, Tatsumi N, Shimizu S, Kanai Y, Nakazawa T., et al. Antiapoptotic function of 17AA(þ)WT1 (Wilms' tumor gene) isoforms on the intrinsic apoptosis pathway. Oncogene. 2006; 25: 4217-4229.
20. Steege A, Fähling M, Paliege A, Bondke A, Kirschner KM, Martinka P, et al. Wilms' tumor protein (-KTS) modulates renin gene transcription. Kidney Internat. 2008; 74: 458-466.
21. Smolen GA, Vassileva MT, Wells J, Matunis MJ & Haber DA. SUMO-1 modification of the Wilms' tumor suppressor WT1. Cancer Res. 2004; 64: 7846-7851.
22. Makki MS, Heinzel T, & Englert C. TSA downregulates Wilms tumor gene 1 (Wt1) expression at multiple levels. Nucleic Acids Res. 2008; 36, 4067-4078.
23. Martínez-Estrada OM, Lettice LOA, Essafi A, Guadix JA, Slight J, Velecela V, et al. Wt1 is required for cardiovascular progenitor cell formation through transcriptional control of Snail and E-cadherin. Nature Genet. 2010; 42: 8993.
24. Vainio S. y Lin Y. Coordinating Early Kidney Development: Lessons From Gene Targeting. Nature Rev Genet. 2002; 3: 533-543.
25. Antonius T, van Bon B, Eggink A, van der Burgt I, Noordam K, van Heijst A. Denys-Drash syndrome and congenital diaphragmatic hernia: another case with the 1097G-A (arg366his) mutation. Am. J. Med. Genet. 2008; 146A: 496-499.
26. Royer-Pokora B, Beier M, Henzler M, Alam R, Schumacher V, Weirich A. y Huff V. Twenty-four new cases of WT1 germline mutations and review of the literature: genotype/phenotype correlations for Wilms tumor development. Am. J. Med. Genet. 2004; 127, 249-57.
27. Robson EJD, He S-J. y Eccles MR. A PANorama of PAX genes in cáncer and development. Nature Rev Cancer. 2006; 6: 52-62.
28. Kawase E, Tanaka K, Honna T, Azuma N. A case of atypical WAGR syndrome with anterior segment anomaly and microphthalmos. Arch Ophthal. 2001; 119: 1855-1856.
29. Tiberio G, Digilio MC, Giannotti A. Obesity and WAGR syndrome. (Letter) Clin Dysmorph. 2000; 9: 63-64.
30. Alders M, Bliek J, vd Lip K, vd Bogaard R. y Mannens M. Determination of KCNQ1OT1 and H19 methylation levels in BWS and SRS patients using methylation-sensitive high-resolution melting análisis. Europ J Hum Genet. 2009; 17: 467-473.
31. Weksberg R, Shuman C. y Beckwith JB. BeckwithWiedemann syndrome. Europ J Hum Genet. 2010; 18: 8-14.
32. Malan V, Chevallier S, Soler G, Coubes C, Lacombe D, Pasquier L, et al. Array-based comparative genomic hybridization identifies a high frequency of copy number variations in patients with syndromic overgrowth. Europ J Hum Genet. 2010; 18: 227-232.
33. Rodríguez-Criado G, Magano L, Segovia M, Gurrieri F, Neri G, González-Meneses A. et al. Clinical and molecular studies on two further families with Simpson-Golabi-Behmel syndrome. Am. J. Med. Genet. 2005; 138A: 272-277.
34. Sakazume S, Okamoto N, Yamamoto T, Kurosawa K, Numabe H, Ohashi Y. et al. GPC3 mutations in seven patients with Simpson-Golabi-Behmel syndrome. Am. J. Med. Genet. 2007; 143A: 1703-1707.
35. Boily G, Saikali Z. y Sinnett D. Methylation analysis of the glypican 3 gene in embryonal tumours. Brit J Cancer. 2004; 90: 1606-1611.
36. Romanelli V, Arroyo I, Rodriguez JI, Magano L, Arias P, Incera I, Gracía-Bouthelier R, Lapunzina P. Germinal mosaicism in Simpson-Golabi-Behmel syndrome. (Letter) Clin. Genet. 2007; 72: 384-386.
37. Barbaux S, Niaudet P, Gubler MC, Grunfeld JP, Jaubert F, Kuttenn F, et al. Donor splice-site mutations in WT1 are responsible for Frasier syndrome. Nature Genet 1997; 17: 467-470.
38. Sonn, G. y Shortliffe, L. M. D.: Management of Wilms tumor: current standard of care. Nature Clin Prac. 2008; 5: 551-560.
Recibido: 9 de octubre de 2010.
Aprobado: 8 de abril de 2011.

