










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Habanera de Ciencias Médicas
versión On-line ISSN 1729-519X
Rev haban cienc méd vol.14 no.5 La Habana sep.-oct. 2015
CIENCIAS CLÍNICAS Y PATOLÓGICAS
Centro Provincial de Genética Médica, Holguín, Cuba
Síndrome de Rothmund-Thomson. Presentación de un caso
Rothmund-Thomson syndrome. Case presentation
Elayne Esther Santana Hernández,I Víctor Jesús Tamayo ChangII y Nancy Cruz MartínezIII
IMáster en Atención Integral al Niño. Especialista Segundo Grado en Medicina General Integral y Genética Clínica. Profesor Asistente. Investigador agregado. esantana@hpuh.hlg.sld.cu
IIMáster en Atención Integral al Niño. Especialista Segundo Grado en Genética Clínica. Profesor Auxiliar. Investigador agregado. vtamayo@hpuh.hlg.sld.cu
IIIEspecialista Primer Grado en Medicina General Integral y Dermatología. Hospital Pediátrico Universitario "Octavio de la Concepción de la Pedraja". Holguín. Cuba. nancy@hpuh.hlg.sld.cu
RESUMEN
Introducción: el Síndrome de Rothmund-Thomson o Poiquilodermia Congénita es considerado una genodermatosis poco frecuente, causada por mutación en el cromosoma 8q.24.3. Clínicamente se caracteriza por degeneración atrófica y pigmentación cutánea anormal, de inicio en la infancia.
Objetivo: describir las características clínicas de un paciente con una afectación hereditaria con múltiples afectaciones dermatológicas.
Presentación de caso: se trata de un paciente masculino de 10 años edad, con cambios atróficos múltiples en cara desde antes de los 6 meses edad y en las extremidades y el tronco, además presentan fotosensibilidad, cabello escaso, retraso marcado del crecimiento.
Conclusiones: resulta importante la realización del diagnóstico precoz de esta entidad por la posibilidad de realizar una prevención secundaria en los pacientes, así como brindar un adecuado asesoramiento genético a la familia.
Palabras clave: poiquilodermia, rothmund-Thomson, genodermatosis, atrofia epidérmica, Síndrome, diagnóstico.
ABSTRACT
Introduction: rothmund-Thomson syndrome or congenital Poikiloderma is considered a rare genetic disorder caused by mutation in the chromosome 8q.24.3. Clinically it is characterized by atrophic degeneration and abnormal skin pigmentation, starting in childhood.
Objective: to describe the clinical features of a patient with an inherited disease with multiple dermatological affections.
Case presentation: this is a male patient 10 years old with multiple atrophic changes in face before 6 months of age and in the limbs and trunk, also present photosensitivity, sparse hair, marked growth retardation.
Conclusions: it is important to conduct early diagnosis of this entity by the possibility of a secondary prevention in patients and provide appropriate genetic counseling to the family.
Key words: poikiloderma, rothmund-Thomson, genodermatosis, epidermica atrophy, syndrome, diagnosis.
INTRODUCCIÓN
El Síndrome de Rothmund-Thomson es una rara afección, su historia se remonta a la Villa de Klein-Walserthal localizada en la Cordillera de los Alpes austriacos, en donde August Von Rothmund (médico oftalmólogo, 1830-1906) reporta en 1868 la asociación familiar de cataratas juveniles, nariz en silla de montar y degeneraciones cutáneas compatibles con atrofia, en tres pacientes con antecedentes de consanguinidad. Medio siglo más tarde Matthew S.1,2 Thomson (médico dermatólogo inglés, 1894-1960) describe en 1923 y 1926 "una enfermedad familiar heredable no descrita" enfatizando los hallazgos cutáneos, a la cual denominó "poiquilodermia congénita" .En la actualidad se considera que los hallazgos de Thomson corresponden a los reportados en los Alpes austriacos por Rothmund mucho tiempo antes.3
Tiene varios sinónimos: Poiquiloderma Congénito, Poiquiloderma Atrófico pero el más aceptado hoy en día es Cutis Marmorata Telangectásica Congénita. Desde el punto de vista cutáneo, se caracteriza por una marcada fotosensibilidad y lesiones de características poiquilodérmicas.4-7
En más de 90% de los pacientes la sintomatología comienza a desarrollarse entre el tercer y sexto mes de vida mediante la presencia de placas eritematosas, de aspecto reticulado, a veces con leve hiperqueratosis, que a lo largo de su evolución, dejan hipo e hiperpigmentaciones residuales, lo cual le confiere a la zona afectada su tan característico aspecto poiquilodérmico. Se caracteriza por degeneración atrófica, pigmentación cutánea anormal y telangiectasias, de inicio en la infancia, asociada a cataratas, fotosensibilidad, estatura corta, anormalidades dentales, ungueales, esqueléticas, osteosarcomas y menos frecuentemente con hipoplasia medio facial, hipermovilidad acral y anemia.Muy raras veces se presentan en el recién nacido.8-10
Las lesiones se describen que comienzan en la cara y se extienden a los glúteos y la superficie extensora de las extremidades. Además presenta una red vascular reticulada profunda, eritematosa violeta, localizada o generalizada presente al nacer. Entre las manifestaciones del área endocrina se encuentran: disfunción tiroidea, talla baja y retraso puberal. Muy pocos casos han sido descritos en la literatura mundial, 6 se totalizan unos 300 casos reportados,7 por lo que consideramos importante, reportar este caso.
OBJETIVO
Describir las características clínicas de las lesiones que presenta desde el nacimiento, considerándose una afectación hereditaria con múltiples afectaciones dermatológicas.
PRESENTACIÓN DEL CASO
Se atiende en consulta provincial de Genodermatosis, en el Centro Provincial de Genética Médica Holguín, un niño con múltiples lesiones en la piel que por su complejidad fue necesario participación de dermatólogos, pediatras, alergistas, endocrinólogos, genetistas clínicos, psicólogos y el trabajador social.
Se presenta un paciente con varias lesiones en la epidermis desde el nacimiento, que según refiere la mamá estas han aumentado de tamaño siendo muy extensa y han cambiado su coloración y textura.
Se discutió el caso en colectivo, se aplicó el método clínico, a través de técnica comparativa y se llegó al diagnóstico de una poiquilodermia congénita o Síndrome de Rothmund-Thomson.
Se le pide consentimiento informado a los padres para examinarlo y fotografiarlo para la publicación del caso. Se confecciona historia clínica genética, se indican exámenes complementarios incluyendo la biopsia de pie de las lesiones; en el proceder se cumplieron con los principios éticos para publicaciones médicas.
Paciente masculino de 10 años de edad, hijo de padres que son primos y sin antecedentes patológicos en la familia de interés, según se muestra en el árbol genealógico es producto de un matrimonio consanguíneo, los padres son primos hermanos. Los antecedentes prenatales y perinatales no son relevantes, nació por parto eutócico, a término de 38 semanas y peso de 2 980 gramos. A los 6 meses de vida comienza a presentar cambios en la piel de la cara de hiperpigmentación con hipopigmentación y atrofia de la piel, esto fue causa de varias consultas de pediatría y dermatología sin definir su diagnóstico.
Al examen físico se evaluó talla, peso para su edad y sexo, que mostró parámetros por debajo del tercer percentil, se diagnosticó como desnutrido. En la piel se observan zonas atróficas en la cara en la región de la frente de forma extensa, así como en las manos y el tronco. Las lesiones son grandes e hiperpigmentadas; de una de estas se toma muestra para biopsia de piel que detalla los cambios atróficos de la epidermis y la dermis. Estas lesiones también son evidentes en otras zonas como el abdomen donde existen cambios de coloración y atrofia marcada, como se puede evaluar en la figura.
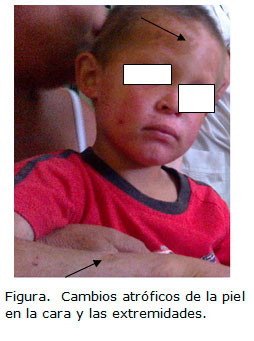
DISCUSIÓN
La Poiquilodermia es un término descriptivo y genérico que se utiliza para denominar una serie de dermatosis que cursa con placas de piel atrófica, salpicadas de telangiectasias, en las que alternan áreas de hiperpigmentación con otras de hipopigmentación que dan a la piel afectada un aspecto moteado.1,2
Este caso corresponde a una poiquilodermia congénita, resultado de prácticas de consanguinidad, igual que lo descritos por otros autores.3-5 El diagnóstico de este síndrome es eminentemente clínico, aunque la biopsia de piel puede ayudar. El cuadro poiquilodérmico de las lesiones en cara, fotosensibilidad, fotodaño marcado, anormalidades oculares y en sus anexos, y el retraso del crecimiento, son patrones muy sugestivos, que se confirman con indagar sobre la práctica de consanguinidad y el patrón de herencia autosómico recesivo del cuadro; sin embargo, ya se han definido dos mutaciones en cromosomas distintos para esta enfermedad como lo descrito en la literatura.5,6
Es una genodermatosis autosómica recesiva que la producen varias mutaciones en el gen RECQL4 localizadas en 8q24, el cual codifica una RecQ DNA helicase5-8, aún no se conoce bien la patogenia de esta enfermedad, pero se sabe que la proteína afectada se encuentra involucrada en el sistema reparación del ADN.
Según la penetración y expresividad génica de este síndrome, se ha reportado una gran variedad de manifestaciones clínicas y anormalidades sistémicas asociadas.7
Entre las manifestaciones clínicas mas frecuentes se encuentran:
• Erupción eritematosa, reticulada y edematosa, que se inicia con más frecuencia en cara, desde los 3 a 6 meses de edad, que luego evoluciona a cambios poiquilodérmicos: atrofia, telangiectasias, hiper o hipopigmentación lineal (muy rara vez ausente), de esta forma se presentó en este paciente.
•Fotosensibilidad en 30 %, que en muchos casos puede producir bulas.
• Lesiones hiperqueratósicas en manos, pies, codos y/o tobillos en 33 % (luego de los 2 años de edad).8
• Distrofia ungueal en 30 %.
Problemas en la dentición en 40%: aumento de la incidencia de caries, microdontia y formas cónicas, lo cual es evidenciable en este niño afectado.
•Cabello fino y escaso que puede progresar a alopecia parcial o total, ocasionalmente ausencia de cejas y pestañas, como se puede apreciar en la figura en este niño dañado.9
•Bajo peso al nacer, con retraso marcado en crecimiento, con estatura proporcionalmente corta.
•Anormalidades esqueléticas en 68 %, que pueden variar desde incapacidades mínimas hasta marcadas: facies características (prominencia frontal), nariz en silla de montar, prognatismo, ausencia o malformación braquimeta carpofalángica, sindactilia, clinodatilia, malformación o agenesia de los huesos del carpo y/o tarso,malformación del radio y primeros dedos de manos, pies pequeños, displasia fibrosa, osteogénesis imperfecta, contracciones de tejidos blandos. Además hiperostosis cortical irregular que simulan condrodistrofias, depresión de la cabeza del radio, osteoporosis e hirpermovilidad acral.
•Anormalidades oculares: cataratas juveniles en 50% (opacidad subcapsular anterior y posterior detectada entre los 3 y 7 años), keratocono, coloboma, estrabismo, ambliopía, discos ópticos oscuros, microftalmia, atrofia del nervio óptico, venas retiniales dilatadas y aneurismales, exoftalmos, atrofia corneal, esclerosis corneal, glaucoma congénito, atrofia corioretineal, fotofobia e hipertelorismo.10
•Malignidades: osteosarcomas en 32 %, carcinoma de células escamosas, Enfermedad de Bowen, fibrosarcoma, linfoma de Hodgkin, carcinoma gástrico y leucemia mielocítica aguda.
•Hipogonadismo en 25 %.
•Anemia y síndrome mielodisplásico.
Otras mucho menos frecuentes como hipoplasia medio facial, calcinosis cutis, queratosis actínicas, retraso mental y disminución del número absoluto y relativo de linfocitos T supresores e IgG4 séricos.
Se debe realizar diagnóstico diferencial con: telangiectasias, Síndrome de Bloom, xeroderma pigmentoso, y otras que guardan relación con el sistema de repación del ADN afectado.
En Cuba no se realiza estudio molecular `para la confirmación de esta enfermedad por lo que resulta un poco complejo llegar al diagnóstico definitivo en ocasiones cuando la biopsia no es concluyente.
El manejo de este paciente fue multidisciplinario; se evaluó por pediatría, oftalmología, ortopedia, neurología, dermatología, genética, especialistas en nutrición y psicología. El tratamiento fue con humectantes, emolientes y protección solar, se realizó vigilancia por malignidades cutáneas y óseas.
El pronóstico está limitado a la aparición de neoplasias; sin embargo, existen muchos reportes de pacientes que superan los 50 años de edad.
La Poiquilodermia congénita es un síndrome de escaso reporte en América; en este momento es el único caso reportado por nuestro Centro Provincial y se mantiene en seguimiento por consulta de genodermatosis.
CONCLUSIONES
Resulta importante la realización del diagnóstico precoz de esta entidad por la posibilidad de realizar una prevención secundaria en los pacientes, así como brindar un adecuado asesoramiento genético a la familia.
REFERENCIAS BIBLIOGRÁFICAS
1. Simon T, Kohlhase J, Wilhelm C, Kochanek M, De Carolis B, Berthold F. Multiple malignant diseases in a patient with Rothmund-Thomson syndrome with RECQL4 mutations: Case report and literature review. Am J Med Genet A. 2010 Jun; 152A(6):1575-9. [Citado 6 Jun 2014]. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/20503338
2. Jin W, Liu H, Zhang Y, Otta SK, Plon SE, Wang LL. Sensitivity of RECQL4-deficient fibroblasts from Rothmund-Thomson syndrome patients to genotoxic agents. Hum Genet. 2008 Jul; 123(6):643-53. [Citado 6 Jun 2014]. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/18504617
3. Piard J, Holder-Espinasse M, Aral B, Gigot N, Rio M, Tardieu M, et al. Systematic search for neutropenia should be part of the first screening in patients with poikiloderma. Eur J Med Genet. 2012 Jan; 55(1):8-11. [Citado18 Jun 2014] Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/21872685
4. Davis T, Tivey HS, Brook AJ, Grimstead JW, Rokicki MJ, Kipling D. Activation of p38 MAP kinase and stress signalling in fibroblasts from the progeroid Rothmund-Thomson syndrome. Age (Dordr). 2013 Oct; 35(5):1767-83. [Citado18 Jun 2014]. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/23001818
5. Fradin M, Merklen-Djafri C, Perrigouard C, Aral B, Muller J, Stoetzel C, Frouin E. Long-term follow-up and molecular characterization of a patient with a RECQL4 mutation spectrum disorder. Dermatology. 2013; 226(4):353-7. [Citado18 Jun 2014]. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/23899764
6. Piard J, Aral B, Vabres P, Holder-Espinasse M, Mégarbané A, Gauthier S. Search for ReCQL4 mutations in 39 patients genotyped for suspected Rothmund-Thomson/Baller-Gerold syndromes. Clin Genet. 2014 Feb 14. [Citado18 Jun 2014]. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/24635570
7. Gupta S, De S, Srivastava V, Hussain M, Kumari J, Muniyappa K, Sengupta S. RECQL4 and p53 potentiate the activity of polymerase and maintain the integrity of the human mitochondrial genome. Carcinogenesis. 2014 Jan; 35(1):34-45. [Citado18 Jun 2014]. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/24067899
8. Monique F. Smeets, DeLuca E, Wall JM, Quach J, Alistair M, et al. The Rothmund-Thomson syndrome helicase RECQL4 is essential for hematopoiesis. J Clin Invest. 2014 August 1; 24(8): 3551–3565. [Citado26 Jun 2015]. Disponible en: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4109528/
9. Larizza L, Roversi G, Volpi L. Rothmund-Thomson syndrome. Orphanet J Rare Dis. 2010; 5: 2. [Citado26 Jun 2015]. Disponible en: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2826297/
10. H Lu, Fang EF, Sykora P, Kulikowicz T, Zhang Y, Becker KG, et al. Senescence induced by RECQL4 dysfunction contributes to Rothmund–Thomson syndrome features in mice. Cell Death Dis. 2014 May; 5(5): e1226. [Citado 26 Jun 2015]. Disponible en: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4047874/
Recibido: 4 de Marzo de 2015.
Aprobado: 2 de Septiembre de 2015.

