










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Habanera de Ciencias Médicas
versión On-line ISSN 1729-519X
Rev haban cienc méd vol.15 no.3 La Habana mayo.-jun. 2016
CIENCIAS CLÍNICAS Y PATOLÓGICAS
Instituto Cubano de Oftalmología "Ramón Pando Ferrer", La Habana, Cuba.
Síndrome de Treacher Collins en una familia cubana. Presentación de caso
Treacher Collins syndrome in a cuban family. Case presentation
Lesly Solís AlfonsoI y Ileana Agramonte CentellesII
IDoctora en Ciencias Médicas. Especialista Segundo Grado en Imagenología. Investigadora Auxiliar. Profesora Auxiliar de la Universidad de Ciencias Médicas de La Habana. leslysa@horpf.sld.cu
IIEspecialista Segundo Grado en Oftalmología. Máster en Ciencias. Profesora Auxiliar de la Universidad de Ciencias Médicas de La Habana. ileanaac@horpf.sld.cu
RESUMEN
Introducción: El Síndrome de Treacher Collins es un desorden genético del desarrollo craneofacial caracterizado por una displasia otomandibular simétrica y bilateral, asociado a diversas anomalías de cabeza y cuello, pero sin afectación de las extremidades. Su expresividad clínica es muy variable, oscilando la incidencia entre 1 en 25 000 y 1 en 70 000 recién nacidos vivos. En la literatura nacional revisada solo se encontraron publicados dos casos: uno, en 1962 y otro, en 2007, pero no se halló ningún reporte sobre familias cubanas afectadas.
Objetivo: Presentar un caso perteneciente a una familia cubana con Síndrome de Treacher Collins.
Presentación del caso: Paciente masculino de 22 años con antecedentes familiares de padre y hermana con Treacher Collins, quien acude a consulta solicitando un posible tratamiento estético. Al examen físico se constató dismorfismo facial típico consistente en: fisuras palpebrales inclinadas hacia abajo, microftalmía, depresión del contorno orbitario, hipoplasia e implantación baja de los pabellones auriculares, micrognatia, hipoplasia de tejidos blandos faciales, y ausencia de pestañas en el tercio externo de los párpados inferiores. Las extremidades, el peso y la apertura bucal del paciente no mostraron alteraciones. Estas malformaciones fueron corroboradas por tomografía computarizada, donde además se detectó aracnoidocele intraselar, y falta de neumatización de celdas mastoideas y seno esfenoidal. El ultrasonido abdominal, el ecocardiograma, la radiografía anteroposterior de tórax y columna resultaron normales.
Conclusiones: Pese a que el síndrome de Treacher Collins es una rara enfermedad congénita, es importante conocerlo para poder hacer un diagnóstico correcto y temprano, que permita ofrecerle al paciente un tratamiento multidisciplinario oportuno.
Palabras clave: Síndrome de Treacher Collins, Síndrome de Franceschetti-Zwahlen-Klein, disostosis mandibulofacial, anomalías craneofaciales, treacle, TCOF 1.
ABSTRACT
Introduction: Treacher Collins syndrome is a genetic disorder of craniofacial development, characterized by a bilateral symmetrical otomandibular dysplasia associated with various abnormalities of the head and neck, with no extremities affection. Its clinical expression is very variable, with a range of occurrence between 1 in 25,000 and 1 in 70,000 live births. Only were found two cases after a review of national published literature, one in 1962 and the other one in 2007; no report of incidence on Cuban families was found.
Objective: To present a case study of a Cuban family with Treacher Collins syndrome.
Case presentation: Male patient 22 year old with father and sister afflicted with Treacher Collins syndrome, arriving to a consulting room seeking for an aesthetic treatment. After physical examination was confirm a typical facial dimorphism consisting of: downward slant palpebral fissures, microphthalmia, orbital edge depression, hypoplasia and ears low implantation, micrognathia, facial soft tissue hypoplasia and absence of eyelashes in the inferior lids external third. Patient extremities, weight and mouth opening showed no abnormalities. These malformations were confirmed by means of a computerized tomography, also was detected an intrasellar arachnoidocele, and lack of pneumatization of mastoid and sphenoid sinus cells. Abdominal ultrasound, echocardiogram, chest and antero-posterior spine radiography were normal.
Conclusions: Although the Treacher Collins syndrome is a rare congenital disease, it is important to know it in order to make a correct and early diagnosis, bringing to the patient an opportune and multidisciplinary treatment.
Keywords: Treacher Collins syndrome, Franceschetti -Zwahlen-Klein syndrome, Mandibulo-facial dysostosis, craniofacial anomalies, treacle, TCOF-1.
INTRODUCCIÓN
Historia, incidencia y etiopatogenia
El Síndrome de Treacher Collins, también denominado Síndrome de Franceschetti-Zwahlen-Klein o disostosis mandibulofacial, fue descrito por primera vez en 1846 por Thompson y, posteriormente, por Berry en 1889. Fue el oftalmólogo inglés E. Treacher Collins, quien detalló sus características principales en 1900. Más tarde, Franceschetti y Klein fueron los que introdujeron la expresión disostosis mandibulofacial, y describieron el perfil de los afectados como similar a la cara de los peces o los pájaros.1,2
Este síndrome es un desorden genético del desarrollo cráneofacial caracterizado por una displasia otomandibular simétrica y bilateral, asociado a diversas anomalías de cabeza y cuello, pero sin afectación de las extremidades. Su trasmisión es autosómica dominante, con penetrancia casi completa (90%) y expresividad clínica considerablemente variable. 3-5
Según la literatura revisada su incidencia estimada oscila entre 1 en 25 000 y 1 en 70 000 recién nacidos vivos.3,6-8
En la mayor parte de los casos (63-93%), este raro trastorno congénito, es causado por una mutación de un gen del cromosoma 5q, al cual se le conoce como TCOF 1. Este gen afectado tiene señales de localización nuclear y nucleolar para la fosfoproteína que lo codifica, la que se nombra treacle. Se piensa que esta proteína ayuda en la etapa de la embriogénesis en el desarrollo del primero y segundo arcos faríngeos, con proliferación, migración y apoptosis aumentada en las células de la cresta neural, entre las semanas 8.5-9.5 de gestación, y altera secundariamente el neuroepitelio craneal 1,3-6,8-11.
Sin embargo, también se han identificado otras mutaciones en este Síndrome, como las de los genes POLR1C (6p21.1) o POLR1D (13q12.2), que codifican para las subunidades I y III de las ARN polimerasas, las que se heredan de forma autosómica recesiva.12,13
Asimismo, en los enfermos de Treacher Collins con microcefalia se han detectado mutaciones en el EFTUD2, 14,15 mientras que en los que desarrollan discapacidades intelectuales se han demostrado anomalías aisladas del CAMK2A o asociadas a las del TCOF1. 11,16 Incluso, se ha reportado un caso de una mutación en el cromosoma 3(q23–q25), la que correspondió a una expresión fenotípica del síndrome más severa.17
Por otro lado, en un experimento donde se exponía a ratones a dosis tóxicas de vitamina A e isotretinoína, se encontró que estos sufrieron de malformaciones semejantes a las expresadas en los pacientes con Treacher Collins.1
Presentar un caso perteneciente a una familia cubana con Síndrome de Treacher Collins.
Dismorfología
Entre las malformaciones craneofaciales más frecuentes se encuentran: hipoplasia simétrica y bilateral de los huesos malares, del reborde infraorbitario (80%), y de la mandíbula (78%) (retrognatia), las que conllevan a una maloclusión dental. Además, es común observar hipoplasia de tejidos blandos a nivel del hueso malar, del reborde orbitario inferior y la mejilla. También se presentan anomalías en las regiones orbitocigomáticas, así como en las articulaciones temporomandibulares que pueden ocasionar limitación de la apertura bucal de gravedad variable. De igual modo, pueden aparecer oblicuidad o inclinación de las fisuras palpebrales hacia abajo (89%), coloboma de párpados inferiores (69%), con ausencia de pestañas en el tercio externo de dichos párpados y microftalmía. Con menos frecuencia el paladar es ojival, y ocasionalmente se constata paladar hendido (28%). A menudo (60%) se descubren anomalías del oído externo, como microtia o anotia, atresia del conducto auditivo externo, y malformaciones de la cadena de huesecillos, que pueden causar una pérdida conductiva de la audición. La detección radiológica de estas malformaciones tiene un papel fundamental para el diagnóstico. 1,3,8,18
Diagnóstico, manejo y pronóstico
El diagnóstico es clínico y puede hacerse en el período prenatal mediante el análisis molecular de muestras de vellosidades coriónicas, y con ecografía, la que revelará el típico dismorfismo facial con anomalías bilaterales del oído. Se pueden realizar estudios genéticos para hallar alteraciones del gen TCOF1, aunque estos no siempre resultan positivos. 1 En este sentido, la resonancia magnética fetal ha evolucionado considerablemente, se convierte en una herramienta incuestionable para la valoración fetal. Ha demostrado ser complementaria a la ecografía, y añade información útil sobre la anatomía orofacial, que permite así una valoración precisa tanto del paladar primario como del secundario.19
El asesoramiento genético es complicado debido a la expresión variable de la enfermedad, por lo que todos los casos deben ser discutidos y manejados por un equipo multidisciplinario, con participación conjunta de las especialidades que se requieran, en función de las manifestaciones clínicas que aparezcan.
Su poca prevalencia y presentación diferente en cada individuo, han dificultado el desarrollo de guías de manejo. Lo más importante es asegurar la integridad de la vía aérea y lograr la alimentación del paciente, sobre todo, durante el período neonatal y la infancia, ya que el Treacher Collins a menudo se asocia con dificultad para la ganancia de peso y apneas obstructivas de leves a graves. 20 El proceder quirúrgico más utilizado y con mejores resultados para este período es la distracción mandibular. También se pueden considerar otros procedimientos, con el fin de preservar la vía aérea, como la traqueostomía, la ventilación no invasiva y las adhesiones labio-lengua. La evaluación por oftalmología, otorrinolaringología y audiología igualmente se debe iniciar entre los 0 y los 36 meses de edad.1
En una segunda etapa, de los 3 a los 12 años de edad, se debe comenzar el tratamiento de ortodoncia, la reparación de los párpados, del paladar o del labio hendido (en caso de que existiesen), y la región cigomática. La otoplastia y otras cirugías de retoque se deben realizar en este lapso.1
En un tercer período, que comprende de los 13 a los 18 años de edad, está recomendado llevar a cabo la cirugía ortognática.21
Por último, el tratamiento siempre debe incluir el apoyo psicológico, tanto a la familia como a los enfermos, no solo por las malformaciones cráneofaciales presentes, sino también por las múltiples intervenciones quirúrgicas a las que necesitarán ser sometidos.
El pronóstico de las formas ligeras y moderadas de la enfermedad es favorable con un tratamiento adecuado. La mayoría de los niños afectados alcanza un desarrollo e inteligencia normales. La atención cuidadosa y precoz garantizará un mejor desempeño escolar.1
PRESENTACIÓN DEL CASO
Se presenta el caso de un paciente masculino de 22 años de edad, con antecedentes familiares de padre y hermana con Síndrome de Treacher Collins, quien acude por primera vez a consulta solicitando un posible tratamiento estético.
Al interrogatorio no se recogieron síntomas de apnea del sueño ni de obstrucción de la vía aérea superior. En tanto al examen físico se constató la existencia de un dismorfismo facial, simétrico y bilateral, característico de este padecimiento, dado por la presencia de fisuras palpebrales inclinadas hacia abajo, microftalmía, depresión del contorno orbitario, hipoplasia e implantación baja de los pabellones auriculares y micrognatia. También se observó hipoplasia de tejidos blandos a nivel del hueso malar, el reborde orbitario inferior y la mejilla, así como ausencia de pestañas en el tercio externo de los párpados inferiores. Finalmente, las extremidades, el peso y la apertura bucal del paciente no mostraron alteraciones.
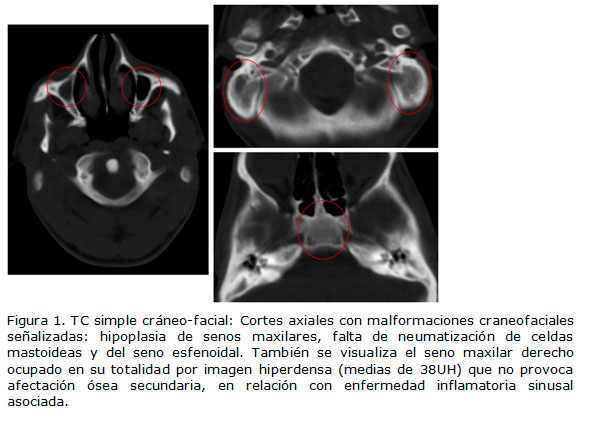
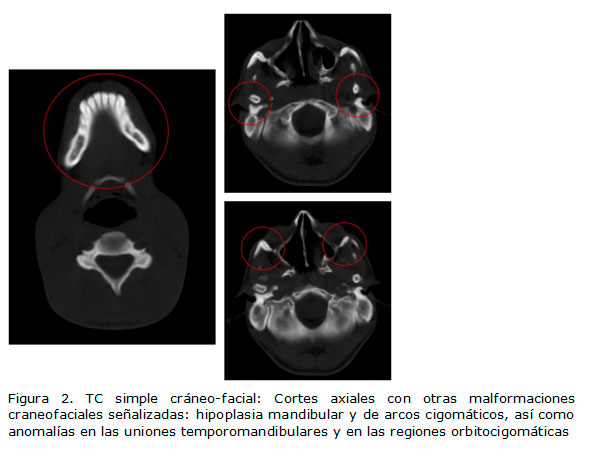
Seguidamente, se le realizó tomografía computarizada (TC) simple de la región cráneofacial, donde se demostró hipoplasia de los senos maxilares, así como ausencia bilateral de neumatización de las celdas mastoideas y el seno esfenoidal. Asimismo, se ratificó la hipoplasia de ambos arcos cigomáticos y de la mandíbula. Además, se comprobó la presencia de anomalías en las uniones temporomandibulares y en ambas regiones orbitocigomáticas. (Figuras 1 y 2).
Por último, se detectó desplazamiento del septum nasal hacia la derecha e imagen de aracnoidocele intraselar. Posteriormente, se le hizo ultrasonido abdominal, ecocardiograma, radiografía posteroanterior de tórax y de columna, los cuales resultaron normales. El paciente fue remitido para tratamiento quirúrgico estético multidisciplinario.
El padre tuvo malformaciones craneofaciales idénticas a las de este caso, pero él sí fue sometido a varias intervenciones quirúrgicas maxilofaciales, que le corrigieron la hipoplasia ósea y los tejidos blandos de forma satisfactoria. La hermana de 12 años, de igual madre, también presenta un dismorfismo facial similar al del paciente que se relata, no obstante, la familia no tiene interés en atenderla por el momento. Tanto el padre como la hermana niegan antecedentes personales de trastornos respiratorios, y al examen físico se comprueban normales las extremidades, el peso y la apertura bucal. En ambos el ultrasonido abdominal, el ecocardiograma, la radiografía anteroposterior de tórax y columna resultaron negativos. Por último, los 3 pacientes muestran un desarrollo e inteligencia normales.
La familia que se describe dio su aprobación para la confección y publicación de este artículo, con la condición de que la información obtenida se utilizará exclusivamente con fines científico-docentes, y que los datos referentes a la identidad permanecieran bajo confidencialidad.
DISCUSIÓN
En 60% de los sujetos afectados por el Síndrome de Treacher Collins los antecedentes familiares son negativos; es decir, se tratan de mutaciones de novo. 3 Sin embargo, el caso analizado sí mostró malformaciones craneofaciales idénticas a las de su padre y hermana.
En esta familia estuvieron ausentes algunas de las malformaciones que más comúnmente conforman este Síndrome, como los colobomas palpebrales inferiores (69%) y labio-paladar hendido (28%), en tanto, se descubrieron otras no descritas como el aracnoidocele intraselar, y la falta de neumatización de celdas mastoideas y el seno esfenoidal; lo que se explica por la gran variabilidad de las manifestaciones clínicas que caracterizan a este padecimiento, o porque simplemente lo acompañen sin guardar relación de tipo causal.
En este paciente, las anomalías detectadas en las articulaciones temporomandibulares no conllevaron a una limitación de la apertura bucal ni a trastornos en la nutrición, y se comprobó su peso dentro de parámetros normales. El resto de la familia tampoco refirió problemas en este sentido.
También se han reportado de forma más inusual la coexistencia de atresia de las coanas, macroglosia, hipertricosis, conexión anómala de venas pulmonares, u otros defectos cardiacos congénitos, atrofia ventricular bilateral, agenesia e hipoplasia renal, fisuras comisurales bilaterales, encondromas y anomalías en la columna.3,6,7,11
Las alteraciones anatómicas traqueo-laríngeas en muchas ocasiones limitan desde el nacimiento la permeabilidad de las vías respiratorias, y constituyen el primer factor agravante en la evolución, lo que incluso puede conllevar a un desenlace fatal, por lo que en el recién nacido afectado por este Síndrome, la permeabilidad de la vía aérea tiene máxima prioridad. Al respecto se han descrito casos de marcado estrechamiento faríngeo, incluso inferior a 1cm, así como de hipoplasia mandibular con retrusión facial muy pronunciada, siendo la combinación de estas anomalías la responsable de la apnea obstructiva y la muerte súbita que puede ocurrir en estos pacientes.3,6,22,23
En Cuba solo se encontraron publicados 2 casos: uno, en 1962 sobre una adulta sometida a tratamiento quirúrgico estético, 24 y otro, en 2007 de una recién nacida fallecida a los 3 meses de edad por severas complicaciones respiratorias. 6 En ambos pacientes no se recogieron antecedentes familiares, por lo que debieron tratarse de mutaciones de novo.
El diagnóstico diferencial de este padecimiento comprende el grupo de las disostosis craneofaciales, donde se incluyen otras raras y complejas enfermedades que comparten características con el Treacher Collins, como el Síndrome de Nager, el de Miller, el de Goldenhar, y la secuencia de Pierre Robin.25
En el Síndrome de Nager las malformaciones cráneofaciales son muy variadas; sin embargo, pueden ser asimétricas como la ausencia o falta de desarrollo de la hemimandíbula inferior, en tanto las anomalías del sistema óseo son múltiples y, sobre todo, de presentación postaxial (extremidades), las que en general abarcan agenesia de uno o varios huesos, estructuras supernumerarias y alteraciones articulares. 26
El Síndrome de Miller se asocia mayoritariamente con afección del sistema nervioso central (lisencefalia y alteraciones craneales), así como a malformaciones óseas, que también son predominantemente postaxiales.26
El Síndrome de Goldenhar es una displasia óculo-aurículo-vertebral, en el que las características clínicas que aparecen casi siempre son unilaterales; consisten las primordiales en: microsomía hemifacial, asociado a defectos vertebrales (hemivértebras, hipoplasia y cotillas anormales), y a malformaciones oculares y auditivas.27
Por último, en la secuencia de Pierre Robin la principal manifestación es la micrognatia, acompañado de paladar hendido (U invertida) y obstrucción de la vía aérea por glosoptosis.28
Idealmente el tratamiento estético debió realizarse en etapas más tempranas de la vida; no obstante, la falta de trastornos respiratorios y nutricionales, así como el desarrollo e inteligencia normales de esta familia, hacen muy favorable el pronóstico de estos pacientes.
CONCLUSIONES
Pese a que el Síndrome de Treacher Collins es una rara enfermedad congénita, es importante conocerlo para poder hacer un diagnóstico correcto y temprano, que permita ofrecerle al paciente un tratamiento multidisciplinario oportuno.
REFERENCIAS BIBLIOGRÁFICAS
1. Leyva JC, Mallarino Restrepo G. Síndrome de Treacher Collins: revisión de tema y presentación de caso. Univ. Méd. 2014;55(1):64-70.
2. Bowornsilp C, Kamonwan J, Prathana C, Palakorn S. Challenges in evaluation, management and outcome of the patients with Treacher Collins syndrome. J Med Assoc Thai. 2011;94(6):S85-90.
3. León Suazo HG, Saucedo Reyes A. Síndrome de Treacher Collins. Reporte de un caso. Rev Mex Pediatr. 2010;77(4):159-163.
4. González B, Henning D, So R, Dixon J, Dixon M, Valdés B. The Treacher Collins syndrome (TCOF 1) gene product is involve in pre-rRNA methylation. Hum Mol Genet. 2005;14(14):2035-43.
5. Teber OA, Gillessen-Kaesbach G, Fischer S, Bohringer S, Albrecht B, Albert A, et al. Genotyping in 46 patients with tentative diagnosis of Treacher Collins syndrome revealed unexpected phenotypic variation. Eur J Hum Genet. 2004;12(11):879-90.
6. Pérez Valdés N, Menéndez García R, Acosta Díaz R, Pérez González Y. Presentación de un caso con Síndrome Treacher Collins. Gaceta Médica Espirituana. 2007;9(3).
7. Eduardo C, Vanier S, Didoni A, Freitas PZ, Carneiro AF, Yoshimoto F. Treacher Collins syndrome with choanal atresia: a case report and a review of its characteristics. Rev Bras Otorrinolaringol. 2005;71(1):107-10.
8. Conte CH, D'Apice MR, Rinaldi F, Gambardella S, Sangiuolo F, Novelli G. Novel mutations of TCOF1 gene in European patients with treacher Collins syndrome. BMC Medical Genetics. 2011;12:125.
9. Beygo J, Buiting K, Seland S. First Report of a Single Exon Deletion in TCOF1 Causing Treacher Collins Syndrome. Mol Syndromol. 2012;2:53-59.
10. Bowman M, Oldridge M, Archer C. Gross deletions in TCOF1 are a cause of Treacher-Collins-Franceschetti syndrome. Eur J Hum Genet. 2012;20:769-777.
11. Vincent M, Geneviève D, Ostertag A, Marlin S, Lacombe D, Martin-Coignard D, et al. Treacher Collins syndrome: a clinical and molecular study based on a large series of patients. Genet Med. 2015;29:1-8.
12. Dauwerse JG, Dixon J, Seland S, Ruivenkamp CA, Van Haeringen A, Hoefsloot LH, et al. Mutations in genes encoding subunits of RNA polymerases I and III cause Treacher Collins syndrome. Nat Genet. 2011;43:20-22.
13. Schaefer E, Collet C, Genevieve D, et al. Autosomal recessive POLR1D mutation with decrease of TCOF1 mRNA is responsible for Treacher Collins syndrome. Genet Med. 2014;16:720-724.
14. Luquetti DV, Hing AV, Rieder MJ. Mandibulofacial dysostosis with microcephaly caused by EFTUD2 mutations: expanding the phenotype. Am J Med Genet A. 2013;161A:108-113.
15. Lines MA, Huang L, Schwartzentruber J. Haploinsufficiency of a spliceosomal GTPase encoded by EFTUD2 causes mandibulofacial dysostosis with microcephaly. Am J Hum Genet. 2012;90:369-377.
16. Vincent M, Collet C, Verloes A. Large deletions encompassing the TCOF1 and CAMK2A genes are responsible for Treacher Collins syndrome with intellectual disability. Eur J Hum Genet. 2014;22:52-56.
17. Manoj K, Rakesh K, Mukesh T, Supriyo G, Jasbir K, Rima D. Cytogenetic and Clinical Assessment of a Family with Treacher Collins Syndrome. Case Reports in Medicine. 2011;2011:1-5.
18. Dixon J, Jones NC, Sandell LL, Jayasinghe SM, Crane J, Rey IP, et al. Tcof1/Treacle is required for neural crest cell formation and proliferation deficiencies that cause craniofacial abnormalities. Proceed Nat Acad Sc. 2006;103(36):13403-8.
19. Zugazaga Cortázar A, Martín Martínez C. Utilidad de la resonancia magnética en el estudio prenatal de las malformaciones de la cara y el cuello. Radiología. 2012;54(5):387-400.
20. Harriet A, Britt Ø, Pamela A, Nina S, Ketil H. Obstructive sleep apnea in Treacher Collins syndrome. Eur Arch Otorhinolaryngol. 2012;269:331-7.
21. Thompson JT, Anderson PJ, David DJ. Treacher Collins syndrome: protocol management from birth to maturity. J Craniofac Surg. 2009;20(6):2028-35.
22. Shah FA, Ramakrishna S, Ingle V, Dada JE, Al Khabori M, Murty PS. Treacher Collins syndrome with acute airway obstruction. Int J Pediatr Otorhinolaryngol. 2000;54(1):41-3.
23. Prado Bernal NY, Villanueva N, Rincón H. Distracción Fuentes R, De la Cuadra JC, Lacassie H, González A. Difficult fiberoptic tracheal intubation in 1 month-old infant with Treacher Collins Syndrome. Brazilian Journal of Anesthesiology. 2015;65(4):1-4.
24. Lescano O. Caso cubano con el Síndrome de Treacher Collins. Rev Cub Cir. 1962;(3):43-49.
25. Prado Bernal NY, Villanueva N, Rincón H. Distracción osteogénica mandibular en síndrome de Nager. Reporte de un caso. Revista Mexicana de Ortodoncia. 2013;1(1):44-53.
26. Vizzuett Martínez R, Villalobos Navarro ID, Rodríguez Araiza D. Disostosis craneofacial. Síndrome de Nager. Rev Esp Méd Quir. 2013;18(1):75-78.
27. Cuesta-Moreno V, Tuesta-Da Cruz O, Silva-Albizuri C. Tratamiento multidisciplinario del Síndrome de Goldenhar. Reporte de caso. Rev. Estomatol Herediana. 2013;23(2):89-95.
28. Sevilla-Paz Soldán RM, Flores-Saavedra S, Rojas-Salazar EG. Síndrome de Pierre Robin: Reporte de un caso. Rev Méd-Cient "Luz Vida". 2013;4(1):58-62.
Recibido: 27 de agosto de 2015.
Aprobado: 30 de marzo de 2016.


{kind=link}
{kind=link}