










My SciELO
 Custom services
Custom servicesServices on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista Habanera de Ciencias Médicas
On-line version ISSN 1729-519X
Rev haban cienc méd vol.16 no.5 La Habana Sept.-Oct. 2017
CIENCIAS CLÍNICAS Y PATOLÓGICAS
Carcinomas renales múltiples como manifestación inicial del Síndrome de Von Hippel Lindau. Presentación de caso
Multiple renal carcinoma as a clinical manifestation of Von Hippel Lindau Syndrome. Case presentation
Lesly Solís AlfonsoI, Ernesto Alemañy RubioII
IDoctora en Ciencias Médicas. Especialista Segundo Grado en Imagenología. Profesora Auxiliar e Investigadora Titular.Instituto Cubano de Oftalmología "Ramón Pando Ferrer". La Habana, Cuba. leslysa@infomed.sld.cu
IIEspecialista Segundo Grado en Oftalmología. Máster en Ciencias. Profesor Auxiliar. Instituto Cubano de Oftalmología "Ramón Pando Ferrer". La Habana, Cuba. ernestoar@infomed.sld.cu
RESUMEN
Introducción: El Síndrome de Von Hippel Lindau es una afección neoplásica multisistémica, heredada de manera autosómica dominante y con alta penetrancia. Su expresividad clínica es muy diversa,oscilando la incidencia entre 1/35000 y 1/36000 nacidos vivos. Esta enfermedad usualmente se diagnostica entre los 20 y 30 años, pero los síntomas pueden aparecer en la infancia. La lesión clínica inicial más común y precoz es el hemangioblastoma de la retina y/o del sistema nervioso central.
Objetivo: Presentar un caso de un paciente con carcinomas renales múltiples como manifestación inicial de un Síndrome de Von Hippel Lindau.
Presentación del Caso: Paciente masculino de 59 años, con antecedentes de salud, quien acude a urgencias por cuadro febril de 3 días de evolución, que fue interpretado como Dengue; se le realizó, dentro de los complementarios, ultrasonido abdominal, donde se descubrió masa sólida a nivel del polo superior del riñón derecho, asociado a existencia de otra en polo inferior de este mismo riñón, así como de 2 más en el contralateral. Además, se encontraron varios quistes pancreáticos y 2 renales izquierdos corticales. El paciente fue intervenido quirúrgicamente; se corroboró el diagnóstico de carcinomas renales de células claras. El examen oftalmológico reveló la presencia de un hemangioblastoma retiniano derecho; en tanto la tomografía computarizada simple de cráneo y la resonancia magnética espinal no mostraron alteraciones.
Conclusiones: Este síndrome es una rara, pero grave afección genética, caracterizada por un alto riesgo de desarrollar enfermedades neoplásicas, lo que hace que sea aún más importante conocerlo, para poder identificar y tratar a tiempo sus temidas complicaciones.
Palabras claves: Síndromede Von Hippel Lindau, carcinomas renales múltiples, hemangioblastomas de retina, quistes renales, quistes pancráticos.
ABSTRACT
Introduction: Von Hippel Lindau Syndrome is a multisystem neoplastic affection, which is inherited as an autosomal dominant trait, with high penetrance. Its clinical expressivity is very diverse, ranging its incidence between 1/35000 and 1/36000 born alive. This disease is usually diagnosed between the 20 and 30 years of age, but its symptoms can appear in childhood. The most common and early initial clinical lesion is the hemangioblastoma of the retina and/or central nervous system.
Objective: To present a case of a patient with multiple renal carcinoma as initial manifestation of Von Hippel Lindau Syndrome.
Case presentation: 59 years old male patient with a history of good health who comes to the Emergency Room because of febrile clinical state of 3 days´ evolution, that was interpreted as dengue. Abdominal ultrasound was included in the complementary studies, in which a solid mass at level of upper pole of right kidney was observed, associated with the existence of another one in lower pole of the same kidney, as well as two others in contralateral. Also, multiple pancreatic cysts and two left cortical renal ones were found. The patient underwent surgery, and the diagnosis of clear cells renal carcinoma was corroborated. The ophthalmological exam revealed the presence of a right retinal hemangioblastoma whereas the plain skull CT-scan, and the magnetic resonance imaging of the lumbar spine did not show any alterations.
Conclusions: This syndrome is a rare, but a serious genetic affection, characterized by a high risk to develop neoplastic diseases; that´s one reason why it is very important to know about it in order to identify, and treat its feared complications in time.
Keywords: Von Hippel Lindau Syndrome, multiple renal carcinoma, hemangioblastoma of the retina, renal cysts, pancreatic cysts
INTRODUCCIÓN
El cáncer de riñón representa la décima causa más común de muerte por cáncer, siendo el carcinoma de células claras su variedad histológica más frecuente (70-80%).1,2 Dentro de los síndromes genéticos asociados con distintos tipos de cáncer renal se incluye al Von Hippel Lindau (VHL), cuyo descubrimiento se debe a Eugen Von Hippel, quien a finales de 1800 describió la angiomatosis retinal congénita. Posteriormente, a principios de 1900, Arvin Lindau enlazó los componentes retinal, cerebral y visceral en una sola entidad con alta morbimortalidad.3-6
El VHL es una afección neoplásica multisistémica, heredada de manera autosómica dominante, y con alta penetrancia (casi completa a los 65 años), estimándose su incidencia en 1 por cada 35 000 a 1 por cada 36 000 recién nacidos vivos, en tanto la prevalencia oscila entre 1 en 31 000 y 1 en 53 000 personas. Sus manifestaciones clínicas son muy diversas y se han descrito más de 40 lesiones en 14 órganos diferentes. El 50% de los pacientes presenta solo una característica, y muy pocos el síndrome completo.1,3,4,7-12
Clínicamente los criterios diagnósticos se dividen en dos, según el paciente cuente o no con historia familiar de la enfermedad. Los mismos se exponen a continuación:1,4,8
1. Una persona que no refiere historia familiar requiere la presentación de dos o más lesiones características, ejemplo:
- Dos o más hemangioblastomas de retina o cerebelo.
- Un solo hemangioblastoma en asociación con una manifestación visceral como: quistes de riñón o páncreas, carcinoma de células renales (antes de los 60 años), feocromocitomas, y, menos comúnmente, tumores del saco endolinfático, cistadenoma papilar del epidídimo o de ligamentos anchos, o tumores neuroendocrinos del páncreas.
2. Una persona con historia familiar solo requiere una de las manifestaciones anteriores.
En la Tabla se resumen las características clínicas de las lesiones clásicas del VHL, previamente descritas.
Este síndrome se produce como consecuencia de una mutación en el gen VHL, situado en el brazo corto del cromosoma 3 (región 3p25-26). Este gen se hereda de forma autosómica dominante, actuando como supresor de tumores, siendo un componente fundamental del complejo proteico que se une y degrada la proteína HIF1 (factor inductor de hipoxia). Cuando se pierde la función del VHL, el HIF1 permanece activo de manera anómala y actúa como estimulador de rutas moleculares carcinogenéticas que activan la proliferación celular.1,3,9,12-16
Teniendo en cuenta las correlaciones genotipo-fenotipo, se han reportado cuatro fenotipos clásicos, basados en el riesgo de feocromocitoma o carcinoma de células renales:1,3
- VHL tipo 1 se caracteriza por un bajo riesgo de feocromocitoma y mutaciones missense (en sentido erróneo) que perturban el plegamiento de la proteína.
- VHL tipo 2 se caracteriza por un alto riesgo de feocromocitoma y otras mutaciones missense, subdividiéndose a su vez en:
. Tipo 2A: bajo riesgo de carcinoma de células renales.
. Tipo 2B: alto riesgo de carcinoma renal.
. Tipo 2C: con un riesgo de feocromocitoma.
El diagnóstico genético molecular, del que se dispone hoy en clínica, permite la confirmación en los enfermos de la mutación del gen VHL, el cribado de los portadores de la mutación entre sus familiares y la detección de portadores, aún sin tumor, para su vigilancia específica. Secundariamente, puede tener valor en el diagnóstico prenatal y en el consejo genético.3
Además de las características anteriores, el patólogo puede sospechar el síndrome cuando coinciden de manera sincrónica o metacrónica:1
- Espectro de lesiones bilaterales: Carcinomas renales de células claras y quistes renales.
- Lesiones quísticas precursoras y formas iniciales de carcinomas renales de células claras.
El diagnóstico de la enfermedad requiere un posterior seguimiento por vida, así como el estudio familiar para detectar nuevos casos. Además, de plantearse un posible embarazo, se deberá brindar consejo genético y considerar la utilización de técnicas de reproducción asistida.12,17
El tratamiento supone un reto de coordinación interdisciplinaria, y debe ir orientado a la evaluación específica de cada órgano susceptible.3 La frecuencia con la que se deberán realizar las exploraciones y exámenes complementarios varía un poco en las diferentes publicaciones, 12,18,19 no obstante, la mayoría coincide en que a los pacientes adultos anualmente se les practicarán los siguientes procedimientos: anamnesis, medición de la tensión arterial, examen físico completo, analítica sanguínea general que incluya cuantificación de catecolaminas y metanefrinas (sobre todo si se detecta hipertensión), fondo de ojo, ecografía abdominal y RM de cerebro y médula espinal con y sin gadolinio. La ecografía abdominal se puede alternar con TC o RM. El paciente debe tener hecha una audiometría basal, y si manifiesta síntomas auditivos se realizará una RM o una TC de oído interno. Si se objetiva alguna otra alteración se completarán los estudios con otras pruebas.3,12 En el caso del examen oftalmológico se recomienda que se inicie en etapas tempranas de la vida (5 años).3
El tratamiento del carcinoma renal requiere la extirpación completa de todas las lesiones sólidas y quísticas, intentando preservar el máximo posible de función renal, debido al alto riesgo de desarrollar tumores a lo largo de la vida; no obstante, en algunos pacientes, podría llegar a ser necesaria la realización de una nefrectomía bilateral. En casos seleccionados podrán emplearse técnicas mínimamente invasivas, como la crioterapia o ablación por radiofrecuencia.3
OBJETIVO
El objetivo de esta investigación es presentar el caso de un paciente con carcinomas renales múltiples como manifestación inicial de un Síndrome de Von Hippel Lindau.
PRESENTACIÓN DEL CASO
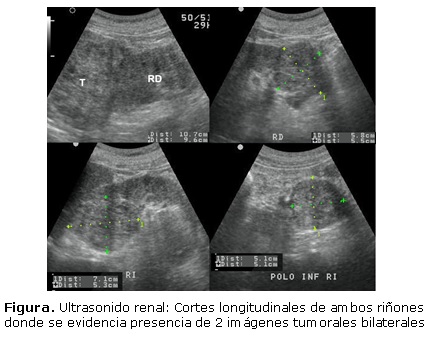
Se presenta el caso de un paciente masculino de 59 años, con antecedentes personales y familiares de salud, quien acude a urgencias por aquejar cuadro febril agudo de 3 días de evolución, que fue interpretado como posible Dengue, y se realiza dentro de los complementarios indicados, ultrasonido abdominal, donde se demostró presencia de masa isoecogénica y heterogénea, a nivel del polo superior del riñón derecho, de contornos irregulares y con vascularidad intralesional, alcanzando 11x10cm en sus diámetros máximos; asociado a la existencia de otra imagen tumoral, de características similares, hacia el polo inferior de este mismo riñón, así como 2 más en el contralateral. (Figura).
Además, se encontraron varios quistes pancreáticos y 2 renales izquierdos corticales. El paciente fue intervenido quirúrgicamente; se corrobó el diagnóstico histopatológico de carcinomas renales de células claras. Posteriormente se le hizo examen oftalmológico (fondo de ojo, tomografía de coherencia óptica y angiografía) y se comprobó la presencia de un hemangioblastoma retiniano derecho. Asimismo, se le realizó tomografía computarizada simple de cráneo y resonancia magnética espinal sin hallarse alteraciones. Durante el interrogatorio refirió tener una hija de 18 años con disminución de la agudeza visual, a la que se exploró y constató presencia de otro hemangioblastoma retiniano derecho. A ella también se le hizo ultrasonido abdominal, tomografía computarizada simple de cráneo y resonancia magnética espinal, los cuales resultaron negativos. Ambos fueron remitidos a consulta especializada de Síndrome de Von Hippel Lindau (VHL). El paciente se encuentra bajo tratamiento médico oncológico.
DISCUSIÓN
El VHL usualmente se diagnostica entre los 20 y 30 años, pero los síntomas pueden aparecer en la infancia; afecta por igual a hombres y mujeres, aunque algunos autores refieren que es más frecuente en el sexo masculino.20 Aproximadamente, 20% de los casos son esporádicos por nuevas mutaciones, siendo en este grupo, considerablemente mayor, la edad media de diagnóstico de tumores.1,12,21 En esta última minoría se incluye el caso que se relata, pues este paciente no refirió antecedentes familiares del síndrome, y antes de los 59 años no tuvo manifestaciones clínicas que permitieran diagnosticar su padecimiento; debutó tardíamente con el hallazgo casual de hipernefromas múltiples.
La lesión clínica más común y precoz es el hemangioblastoma de la retina (70%) y/o del sistema nervioso central (cerebelo, médula espinal y tronco cerebral), que aumenta su frecuencia con la edad.3,4,8,22-24 De hecho, esta lesión retiniana estuvo presente en los dos casos que se describen, pero pese a ocasionar disminución de la agudeza visual, no fueron motivo de consulta médica previa.
Los hemangiomas capilares retinianos representan la variedad histopatológica más usualmente encontrada; no obstante, el hemangioma yuxtapapilar ha sido descrito en 11-15% de los afectados por este síndrome. La pérdida visual asociada puede deberse a varias causas, entre ellas, los fenómenos exudativos (25%) o traccionales (9%), el desarrollo de membranas epirretinianas y la hemorragia vítrea. Incluso se ha demostrado que la función retiniana de los pacientes con enfermedad de VHL puede estar dañada aún en ausencia de hemangioblastomas. El diagnóstico de hemangioblastoma retiniano obliga a completar el estudio sistémico en busca de otras lesiones viscerales que puedan limitar la supervivencia, como el carcinoma renal.8
La afectación del aparato genitourinario en el VHL se limita básicamente a los riñones, las glándulas suprarrenales y los epidídimos. Estas manifestaciones suelen ser precoces, multifocales y bilaterales.3
Entre las lesiones renales que pueden aparecer se reportan quistes, adenomas, angiomas y carcinomas. Los quistes se observan en casi las 2/3 partes de los afectados, y suelen ser pequeños, múltiples, corticales y bilaterales. La apariencia abarca desde los quistes simples (Bosniak I) hasta los complicados (Bosniak IV). El epitelio displásico y claramente maligno que puede revestir las paredes de estos quistes, así como su potencial maligno son difíciles de establecer mediante técnicas de imagen convencionales. Además, la aparición de quistes puede preceder a los tumores en 5 años o más. Estas características han llevado a algunos autores a definir este tipo de quistes como lesiones precursoras de tumores renales sólidos en el VHL. En este sentido, los portadores del síndrome tienen 40% de riesgo de desarrollar cáncer de riñón, correspondiendo en su mayoría a carcinomas de células claras, que suelen ser multifocales y bilaterales hasta en 75% de los casos, con una edad media de aparición cercana a los 40 años, siendo esta la principal causa de mortalidad en el VHL.1,3,4,25 Dichas lesiones urológicas a menudo permanecen asintomáticas durante largos intervalos de tiempo, lo que retrasa su detección temprana,4 como ocurrió en el paciente que se reporta.
El feocromocitoma adrenal afecta a 10-20% de los pacientes, y las lesiones del epidídimo incluyen quistes simples y cistadenomas papilares.3,26
Ong KR y su grupo describieron la media de edad al diagnóstico, así como la frecuencia lesional, en 573 casos con cinco lesiones mayores del VHL, siendo las mismas: hemangioma capilar retiniano 25 años (73%), hemangioblastomas cerebelares 29.9 años (57%), carcinoma de células renales 39.7 años (35%), hemangioblastoma espinal 33.3 años (25%) y feocromocitoma 24 años (20%).27
El páncreas, al igual que el riñón es comúnmente afectado en el VHL, siendo en la mayor parte de los casos por quistes simples, los que pueden constituir la primera manifestación del síndrome cuando se realiza una exploración de rutina. La frecuencia de estas lesiones según las necropsias es cerca de 72%. Los quistes generalmente son pequeños, pero en ocasiones pueden reemplazar u ocupar la totalidad del órgano, en cuyo caso se ha descrito la posible asociación con Diabetes Mellitus. Otras lesiones pancreáticas que han sido asociadas al VHL incluyen el carcinoma pancreático, carcinoma de la Ampolla de Vater, hemangioblastoma, adenoma microquístico y tumores no funcionales de las células de los islotes.26,28,29
En este sentido Levine, et al, tras estudiar mediante TC de abdomen a 31 integrantes de tres familias con VHL, encontraron 13 con quistes renales, 4 con carcinomas renales, 5 con quistes pancreáticos y 2 con feocromocitomas.26
Se quiere aclarar que en la literatura consultada no se encontró referencia de otros casos con carcinomas renales como forma de presentación del Síndrome de Von Hippel Lindau.
CONCLUSIONES
El Síndrome de VHL es una rara, pero grave afección genética, caracterizada por un alto riesgo de desarrollar enfermedades neoplásicas, lo que hace que sea aún más importante conocerlo, para poder identificar y tratar a tiempo sus temidas complicaciones. Lo ideal sería siempre poder lograr el diagnóstico precoz, pero de resultar un hallazgo incidental, como es el caso que aquí se presenta, al menos permitiría adoptar acciones tempranas sobre los familiares en riesgo, lo que garantizaría el tratamiento oportuno y un mejor pronóstico.
REFERENCIAS BIBLIOGRÁFICAS
1. Sanz-Ortega J, Olivier C, Pérez Segura P, Galante Romo I, San José Mansó L, Sáez M. Cáncer de riñón hereditario. Actas Urol Esp. 2009; 33(2):127-133.
2. Solís Alfonso L, Suárez Pría SD. Metástasis amigdalina como presentación de un carcinoma de células renales. Actas Urol Esp. 2015; 39(4):269-270.
3. Cabrera López C. Enfermedades genéticas tumorales renales. Nefrología Sup Ext. 2011; 2(1):97-101.
4. Rojas Barrantes EI. Enfermedad de Von Hippel Lindau. Revista Médica de Costa Rica y Centroamérica. 2013; LXX (605):181-184.
5. JM Torpy MD, C Lynm MA, R Golub MD. Cáncer de riñón. JAMA. 2011; 306(2):226.
6. Acevedo PA, Raichholz G, La Roca C, Rovere L, Stoisa D, Staffieri R. Tumores renales bilaterales. Anuario Fundación Dr. JR Villavicencio. 2007; XV:31-34.
7. Wen-Chung W, Mei-Hua T, Hui-Ju Ch, Wei-Fang H, Yen-Chein L. Two single nucleotide polymorphisms in the Von Hippel-Lindau tumor suppressor gene in Taiwanese with renal cell carcinoma. BMC Research Notes. 2014; 7:638-46.
8. Salazar R, González-Castaño C, Rozas P, Castro J. Hemangioma capilar retiniano y enfermedad de Von Hippel-Lindau: implicaciones diagnósticas y terapéuticas. Arch Soc Esp Oftalmol. 2011; 86(7):218-221.
9. Gossage L, EV Pires D, Olivera-Nappa A, Asenjo J, Bycroft M, Blundell TL, et al. An integrated computational approach can classify VHL missense mutations according to risk of clear cell renal carcinoma. Human Molecular Genetics. 2014; 23(22):5976-5988.
10. De Arteaga J, Colla R, Metrebian S, Ruggieri M, Alvarellos T, Rossi N, et al. Enfermedad de Von Hippel Lindau, tratamiento del cáncer renal y complicaciones en diálisis crónica. Mecanismos moleculares de producción del tumor. Experiencia Médica. 2009; 27(2):54-58.
11. López JI, Ugalde A, Zhou M. Carcinomas renales con células claras. Rev Esp Patol. 2008; 41(3):169-182.
12. Etxeberria-Lekuona D, Casas Fernández de Tejerina JM, Zazpe I, Teijeira Sánchez L. La enfermedad de Von Hippel-Lindau y el papel del internista en las enfermedades raras. Rev Clin Esp. 2012; 212(8):e59-61.
13. Hernández Fernández RA. Fundamentos moleculares de la enfermedad de Von Hippel Lindau. Rev Cubana Invest Biomed. 2010; 29(2):262-273.
14. Sutapa S, Gourish M, Eun Ju H, Da Woon H, Shamit KD, Seethalakshmi L, et al. Von Hippel-Lindau gene product directs cytokinesis: a new tumor suppressor function. J Cell Sci. 2011; 124:2132-2142.
15. Shagufta K, Sadia A, Sadaf F, Saba S, Asad Shahzad H, Gauhar S, et al. Unique molecular alteration patterns in Von Hippel-Lindau (VHL) gene in a cohort of sporadic renal cell carcinoma patients from Pakistan. Mutation Research. 2014; 763:45-52.
16. Xin M, Donglai S, Hongzhao L, Yu Z, Xiangjun L, Qingbo H, et al. MicroRNA-185 inhibits cell proliferation and induces cell apoptosis by targeting VEGFA directly in von Hippel-Lindau? inactivated clear cell renal cell carcinoma. Urologic Oncology. 2015; 33(4):169.e1-169.e11.
17. Obradors A, Fernández E, Rius M, Oliver-Bonet M, Martínez-Fresno M, Benet J, et al. Outcome of twin babies free of Von Hippel-Lindau disease after a double-factor preimplantation genetic diagnosis: monogenetic mutation analysis and comprehensive aneuploidy screening. Fertil Steril. 2009; 91(933):e1-7.
18. Lonser RR, Glenn GM, Walther M, Chew EY, Libutti SK, Lineham WM. Von Hippel-Lindau disease. Lancet. 2003;361:2059-2067.
19. Hes FJ, Höppener JW, Luijt RB, Lips CJ. Von Hippel-Lindau disease. Hered Cancer Clin Pract. 2005; 3:171-178.
20. Castillo Salgado C, Vargas Rivadeneira E, Madera Grijalva A. Hemangioblastoma de fosa posterior asociado a enfermedad de Von Hippel Lindau. Rev. Ecuatoriana de Neurología. 2009; 18(1-2).
21. Humera S, Shahnaz Imdad K, Shahzad A, Naila T. Expression of Von Hippel-Lindau (VHL) gene mutation in diagnosed cases of renal cell carcinoma. Pak J Med Sci. 2014; 30(4):880-885.
22. Merrill MJ, Edwards NA, Lonser RR. Hemangioblastoma associated mast cells in von Hippel-Lindau disease are tumor derived. Blood. 2014; 121(5).
23. Shively SB, Falke EA, Jie L, Tran MGB, Thompson ER, Maxwell PH, et al. Developmentally arrested structures preceding cerebellar tumors in von Hippel–Lindau disease. Modern Pathology. 2011; 24:1023-30.
24. Zhengping Z, Frerich JM, Huntoon K, Yang CH, Merrill MJ, Ziedulla A, et al. Tumor derived vasculogenesis in Von Hippel-Lindau disease-associated tumors. Scientific Reports. 2014; 4:4102-108.
25. Bautista Núñez D, López Caballero I, Santaella Torres F, Sánchez Martínez LC. Enfermedad de Von Hippel Lindau: Tumor renal bilateral, reporte de un caso y revisión de la literatura. Bol Coleg Mex Urol. 2012; XXVII(3):134-138.
26. Levine E, Collins DL, Horton WA, Schimke RN. CT Screening of the Abdomen in Von Hippel-Lindau Disease. AJR. 1982; 139:505-510.
27. Ong KR, Woodward ER, Killick P. Genotype-phenotype correlations in von Hippel-Lindau disease. Hum Mut. 2007; 28:143-149.
28. Taouli B, Ghouadni M, Correas JM, Hammel P, Couvelard A, Richard S, et al. Spectrum of Abdominal Imaging Findings in Von Hippel-Lindau Disease. AJR. 2003; 181:1049-1054.
29. Weisbrod AB, Kitano M, Thomas F, Williams D, Gulati N, Gesuwan K, et al. Assessment of Tumor Growth in Pancreatic Neuroendocrine Tumors in von Hippel Lindau Syndrome. Journal of the American College of Surgeons 2014; 218(2):163-169.
Recibido: 5 de diciembre de 2016.
Aprobado: 3 de Agosto de 2017.


{kind=link}
{kind=link}