










 Serviços customizados
Serviços customizados
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkIntroducción
Las enfermedades de la motoneurona (MN) son un grupo heterogéneo de trastornos en los que la pérdida selectiva de función de las motoneuronas su periores y/o de las motoneuronas inferiores deriva en una alteración del control del movimiento voluntario del sistema nervioso. La enfermedad más frecuente es la esclerosis lateral amiotrófica (ELA) adquirida o esporádica de debut tardío, trastorno combinado de motoneurona superior e inferior.1,2,3,4,5,6 Los análisis genéticos han contribuido a esclarecer la patogenia de algunas enfermedades de neuronas motoras como la que nos ocupa.1,2,3,5,6,7
La enfermedad es inexorablemente progresiva y conduce a la muerte por parálisis respiratoria, la supervivencia media es entre tres y cinco años. El deterioro progresivo de los pacientes con ELA genera un gran impacto sobre su calidad de vida. Su incidencia aumenta a partir de los 50 años. Se han publicado algunos casos raros de estabilización o incluso de regresión de una ELA. En la mayor parte de las sociedades hay una incidencia de uno a tres casos nuevos por 100 000 habitantes y una prevalencia de tres a seis por 100 000 habitantes.
Se han descrito focos endémicos de mayor prevalencia en el Pacífico occidental (en determinadas regiones de Guam y Papúa-Nueva Guinea). En Estados Unidos y Europa, los varones se afectan con una frecuencia algo mayor que las mujeres. Estudios epidemiológicos han señalado algunos factores de riesgo para esta enfermedad que incluyen la exposición a plaguicidas e insecticidas, tabaquismo y servicio en el ejército. Las susceptibilidades genéticas son los únicos factores de riesgo probados, aunque los efectos del atletismo/ejercicio físico y otros posibles factores ambientales se han considerado.1,2,3,4,5,6
El proceso de degeneración neuronal en la ELA es complejo y se invocan varios mecanismos relacionados. Se plantea que cada vez son mayores las evidencias que apoyan que la ELA no es una enfermedad sino un síndrome clínico que se caracteriza por una degeneración de ambas motoneuronas y que comparten una sintomatología clínica característica.8
La sobrevida es altamente variable, con un amplio rango desde unos pocos meses hasta varios años. Registros prospectivos de la población reportan un rango de un año de mortalidad después del diagnóstico de un 22 % hasta un 34 %.9,10 La identificación temprana de las variantes potencialmente malignas de ELA fue esencial para que los pacientes tomaran decisiones vitales en correspondencia con los neurólogos para decidir a favor o en contra de la posibilidad de realizar intervenciones invasivas como gastrostomía o ventilación mecánica.
Estudios poblaciones han identificado diferentes factores pronósticos de sobrevida entre los cuales se incluyen factores demográficos (edad de inicio, sexo), factores clínicos (tipo de inicio, forma clínica, rango de progresión de la enfermedad), y factores relacionados con la nutrición o respiración.9,10,11,12,13,14
La ELA no es una enfermedad curable. Todos los pacientes mueren a corto plazo según reportes internacionales. El intervalo desde el diagnóstico hasta la muerte no está definido, es multifactorial y variable de paciente a paciente. En general, se establece un margen de 12 hasta 48 meses a partir del diagnóstico,14,15,16,17) por lo que es necesario conocer realmente la sobrevida y las causas directas del fallecimiento en nuestro medio. ¿Qué factores pueden modificarla? ¿Viven más los pacientes cubanos que los de otros países? ¿Qué parámetros clínicos influyen sobre la sobrevida? Las respuestas a estas interrogantes nos permitirían comparar los resultados de otros autores y ver el comportamiento de la enfermedad en la población cubana. No existen estudios publicados en Cuba que aborden la sobrevida de estos pacientes.
Por tanto, el objetivo del presente estudio es caracterizar la sobrevida de los pacientes con diagnóstico de esclerosis lateral amiotrófica partiendo de factores relacionados con su comportamiento clínico en el Instituto Nacional de Neurología y Neurocirugía “Dr. José Rafael Estrada González” de La Habana, Cuba.
Material y Método
Se realizó una investigación descriptiva y retrospectiva de una serie de 147 casos de pacientes diagnosticados con ELA, por la confirmación clínica, neurofisiológica e imágenes, atendidos en la consulta multidisciplinaria en el periodo de octubre de 2005 a octubre de 2015.El universo estuvo constituido por los 147 pacientes de la serie pertenecientes a la provincia La Habana, los cuales atendiendo a los Criterios Diagnósticos de ESCORIAL y AWAJI18 se clasificaron como ELA definida, de ellos hubo 110 fallecidos y 37 continúan vivos.
Como fuentes secundarias para la recolección de la información fueron utilizadas una base de datos existente (serie) en el Instituto de Neurología y Neurocirugía, las historias clínicas del archivo y los certificados de defunción archivados en el Ministerio de Salud Pública.
Se confeccionó una base de datos para la presente investigación en Microsoft Excel con las variables que fueron objeto de estudio: grupos de edades: menos de 30 años, 31 a 40 años, 41 a 50 años, 51 a 60 años, 61 a 70 años y 70 años y más; sexo: femenino y masculino; color de la piel: blanca, negra, mestiza, amarilla; ocupación.
Además se estudiaron: nivel de escolaridad: primario, medio y superior o universitario; antecedentes patológicos personales de hipertensión arterial, cardiopatía isquémica, asma bronquial, diabetes mellitus y eventos cerebrovasculares; antecedentes familiares de la enfermedad; factores de riesgo como exposición a sustancias tóxicas, radiaciones, ejercicios físicos intensos y vida militar; hábitos tóxicos entre los que se consideraron tabaquismo, alcohol y café; síntomas de inicio de la enfermedad; forma clínica: espinal o bulbar; resultados del tratamiento con riluzol en los pacientes a los cuales se les aplicó; tiempo de sobrevida desde el diagnóstico hasta el fallecimiento; lugar del fallecimiento: hogar o institución hospitalaria y causa directa del fallecimiento por certificado de defunción.
Dentro del análisis estadístico se utilizaron como medidas de resúmenes las frecuencias absolutas y relativas. Se realizó la prueba chi-cuadrado de independencia de Pearson (X2) con corrección de Yates para la asociación de variables. Se utilizó un nivel de significación del 5 %. Se calculó la sobrevida interrelacionando las variables estudiadas aplicando la curva de Kaplan Meier. Se aplicó medias y medianas con un intervalo de confianza del 95 % para el límite inferior y superior. Se utilizó el análisis univariante en variables significativas y se incorporaron al análisis multivariante después de analizar las tablas de supervivencia. Se utilizó la prueba chi-cuadrado para buscar la relación entre sexo y forma clínica.
Se respetó el principio a la confidencialidad de la información de la base de datos revisada y las historias clínicas. Fue aprobada la investigación por el Consejo científico y el Comité de ética de las investigaciones.
Resultados
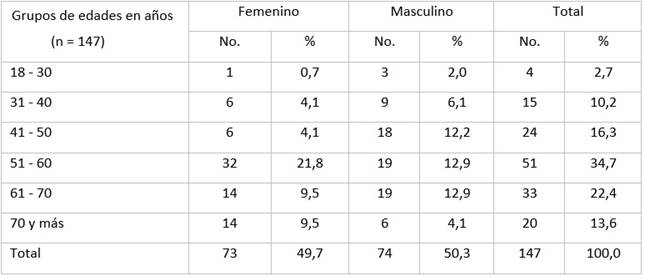
La mayor frecuencia de la enfermedad por grupos de edades estuvo entre 51 y 60 años, seguidos del grupo de 61 a 70 años. Entre ambos representan el 57 % del total de los casos, o sea, más de la mitad. Es de señalar que la frecuencia por grupos de edades se eleva a partir de los 51 años lo que representa el 71 % del total. (Tabla 1).
Según los CRITERIOS DE ESCORIAL y AWAJI,18 143 pacientes se clasificaron como ELA Definida, representando el 97,3 % de los casos. En 4 pacientes no fue concluyente.
En la Tabla 2 se recogen algunas variables sociodemográficas del estudio. El mayor número de pacientes eran de color de la piel blanca, de procedencia urbana, sin antecedentes patológicos o comorbilidades y cuyos hábitos tóxicos fueron tabaco, alcohol y café. En cuanto a la ocupación, 108 eran trabajadores, vinculados fundamentalmente a la producción, la agricultura y los servicios. La mayoría no tenía antecedentes patológicos personales.
Las ocupaciones que demandan mayor actividad física se relacionaron con la aparición de la enfermedad. A algunas de estas ocupaciones se asoció manipulación de sustancias tóxicas, exposición a pesticidas y a metales pesados, traumatismos ocupacionales, estrés, campos electromagnéticos, ejercicios intensos. Muchos de estos pacientes practicaban además deportes, tales como judo, kárate, pelota y boxeo, en los que existe una actividad física intensa. En el caso de las mujeres predominaron las amas de casa.
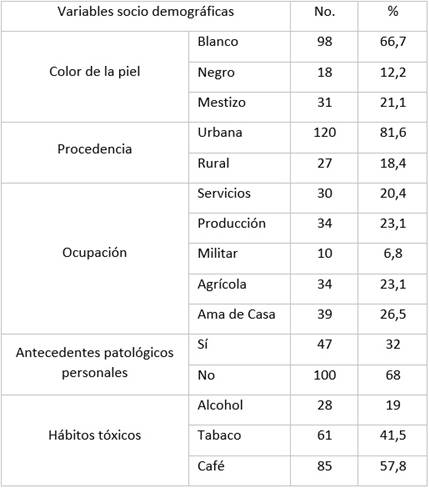
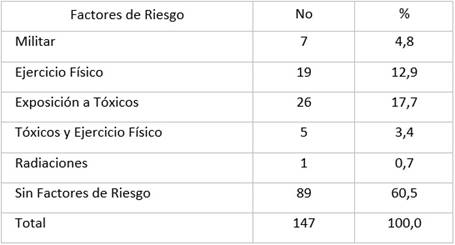
Los síntomas de inicio recogidos en las historias clínicas en el momento del diagnóstico se ilustran en la Tabla 3. La tercera parte de los pacientes refirieron debilidad de los miembros superiores, así como disartria. En el resto se destaca la debilidad de los miembros inferiores, dificultad respiratoria, fasciculaciones, atrofia muscular y debilidad hemicorporal.
Al identificar en el diagnóstico la forma clínica de la enfermedad, obtuvimos los resultados que se muestran en la Tabla 4. Predominó la forma espinal en la mayoría de los pacientes y en hombres. La forma clínica bulbar predominó en mujeres.

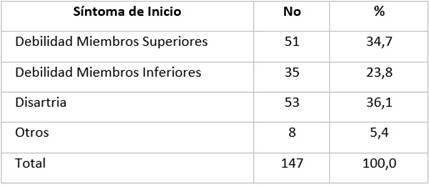
De los factores de riesgo recogidos en la literatura, lo más frecuente se recoge en la Tabla 5. En la mayoría de los pacientes del estudio no pudo identificarse factores de riesgo (60,5%). En el resto se destaca la exposición a tóxicos, actividad física intensa, militares, exposición a radiaciones y asociación entre ellos. Muy pocos pacientes tenían fenotipo y genotipo identificados por lo que no se tomó en cuenta.
De los pacientes del estudio, 44 murieron en su domicilio y 103 fallecieron en unidades hospitalarias que representa el 70 %. No pudo identificarse la causa directa de muerte pues el cierre de los certificados de defunción, tanto en la causa básica como en la directa decían “Enfermedad Neuromuscular”.
Con respecto a la escolaridad de los pacientes, 12 tenían nivel escolar primario para un 8,2 %; el resto tenía nivel escolar entre medio superior y superior para un 91,8 %.
La comorbilidad o APP identificados fueron: la hipertensión arterial, la diabetes, enfermedad cerebrovascular isquémica, neoplasias, hepatitis C, traumatismo craneal, asma bronquial y la cardiopatía isquémica. En los APF hubo casos de la enfermedad en una misma familia.
Análisis de la supervivencia.
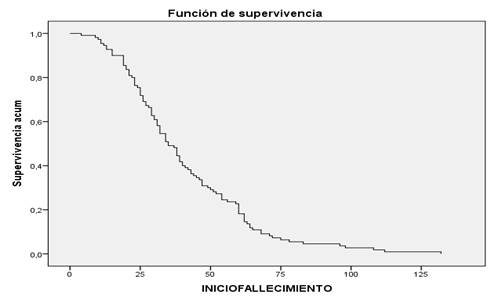
El 80 % de los pacientes falleció en los primeros dos años (24 meses) después del diagnóstico. Entre los 40 y 65 meses (3 y 5 años y medio) fallecieron 19 pacientes. Los restantes 11 pacientes alcanzaron una sobrevida mayor de 6 y medio años (80 meses), llegando muy pocos a los 10 años (120 meses). 37 pacientes aún vivían al cierre del 2015. (Figura 1) (Tiempo de sobrevida primaria, acumulado a los 10, 20 y 30 meses. Acumulado al término del estudio. La pendiente de la curva representa cuantos mueren por mes de progresión de la enfermedad, en cada periodo. Esto es útil para definir la probabilidad de muerte en cada etapa de la enfermedad.)
Las curvas de supervivencia según género fueron similares en las etapas tempranas y tardías de la enfermedad. En las etapas intermedias hay mayor mortalidad en las mujeres.
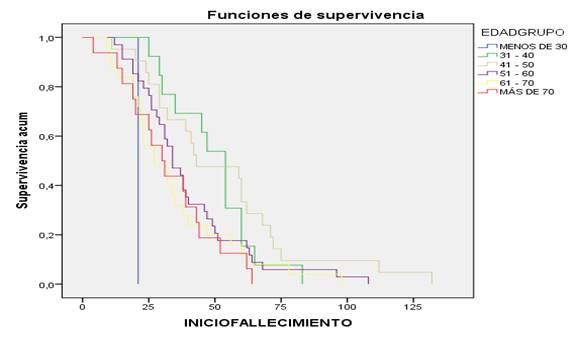
Al observar la curva de supervivencia por grupos de edades (Figura 2), resalta que en los menores de 30 años la supervivencia fue menor a partir del diagnóstico, menos 2 años (24 meses) relacionado con formas más graves de la enfermedad y que tenían predisposición genética familiar. Entre 31 y 40 años y mayores de 70 años fallecieron entre los 2 y 6 años. Los diagnosticados entre 41 y 50 años lograron sobrevivir 10 años. El menor grupo de pacientes logra sobrevivir más de 80 meses. En los que mantenían comorbilidad asociada, la supervivencia alcanzó solo 4 años (48 meses); los que no tenían comorbilidad asociada alcanzaron mayor supervivencia y algunos hasta 10 años.
Entre los hábitos tóxicos, el tabaquismo y la ingestión de café fueron los más frecuentes. Este último no descrito en la literatura como factor de riesgo. Solo el 19 % de los casos consumía alcohol. El análisis de la supervivencia no demostró influencia significativa.
Hubo predominio urbano en la procedencia, lo cual resulta lógico pues el 75 % de la población cubana reside en áreas urbanas. Además el estudio correspondió a los casos de La Habana. Al analizar la supervivencia desde el diagnóstico de la enfermedad hasta el fallecimiento un mayor número de pacientes de procedencia urbana logran mayor sobrevida.
A pesar de predominar la enfermedad en los pacientes de la serie de color de piel blanca, el análisis de supervivencia realizado demostró mayor sobrevida en algunos pacientes de piel negra que supera los 10 años (120 meses). En el resto entre 8 y 10 años (96 y 120 meses).
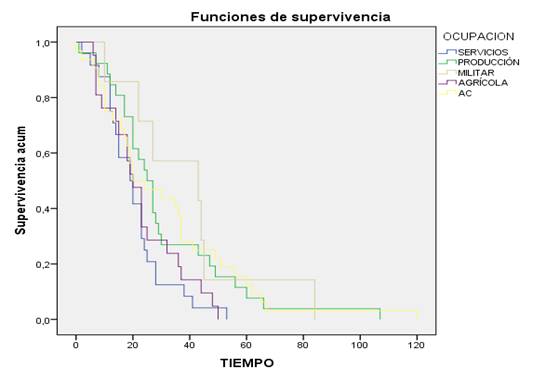
La mayor supervivencia atendiendo a la ocupación estuvo en los trabajadores de la producción, los militares y las mujeres amas de casa. (Figura 3).
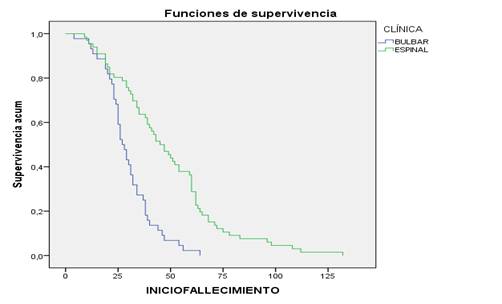
En la curva de supervivencia atendiendo a la forma clínica, tuvieron mayor tiempo de supervivencia los pacientes con forma clínica espinal, algunos alcanzaron hasta unos 10 años (120 meses) (Figura 4)
La supervivencia por factores de riesgo, en el paciente expuesto a radiaciones fue menor, no llegando a 2 años (24 meses) a partir del diagnóstico. El resto de los pacientes, independientemente del tipo de factor de riesgo, la mitad falleció antes de los 2 años (24 meses) y el resto lo hizo entre los 2 y 5 años (24 a 60 meses). Ningún paciente con factores de riesgo sobrevivió más de 60 meses. Un pequeño número sin factores de riesgo logró sobrevivir hasta unos 10 años (120 meses).
Recibieron tratamiento con Riluzol 45 pacientes y no lo recibieron 65. El riluzol no prolonga sobrevida en el análisis de las curvas de supervivencia de esta serie. Ambos grupos tuvieron una supervivencia máxima entre 8 y 10 años (96 a 120 meses).
Discusión
La esclerosis lateral amiotrófica es una enfermedad degenerativa de curso progresivo. La certeza en el diagnóstico se obtiene durante la autopsia, aunque existe un consenso general de que los pacientes pueden ser diagnosticados en vida a pesar de que es difícil en los inicios de la enfermedad, por lo cual en nuestra investigación partimos del momento del diagnóstico y retrospectivamente en las fuentes de datos aparecen recogidos síntomas de inicio, pero no con un tiempo preciso. Es posible que el inicio haya ocurrido un tiempo antes. El diagnóstico es esencialmente clínico teniendo en cuenta los criterios de ESCORIAL, con soporte en RMN, estudios electrofisiológicos y electromiográficos, y genéticos.1,2,3,5,6,18
El presente estudio no ha sido realizado en Cuba hasta el presente por lo que no tenemos datos nacionales para comparar los resultados. La literatura revisada señala que la enfermedad es más frecuente en hombres que en mujeres y su máxima aparición está a partir de los 60 años; otros mencionan que a partir de los 50.1,2,3,4,5,6,9) En nuestra serie, el mayor número de casos se encontró a partir de los 51 años.
Resultó significativo estadísticamente la presencia de la enfermedad en mujeres a partir de los 51 años, en las que predominó la forma bulbar. En los menores de 50, hubo mayor frecuencia en los hombres. Este resultado puede estar relacionado con las formas clínicas de presentación, en las que predominó la espinal que tiene mejor pronóstico y mayor supervivencia. Además, pueden existir diferencias en cuanto al fenotipo y genotipo de los más jóvenes que influyan en la menor sobrevida.
Existen diferencias entre los fenotipos/genotipos de los pacientes que fallecen más tempranamente según la literatura. Los casos de la serie estudiada, la mayoría no tenían la caracterización fenotípica/genotípica en las historias clínicas, lo que no permite concluir al respecto.
Estudios consultados realizados en Italia19 y en Latinoamérica20 aportan diferencias. En Italia la proporción hombre-mujer es prácticamente 1:1. En la investigación de Latinoamérica predominaron las mujeres; en ambos prevaleció la forma bulbar. En este último estudio señalan la posible influencia en los resultados del nivel socio económico, costumbres culturales y ocupaciones en las que se destacan las labores agrícolas con esfuerzos físicos. En nuestra serie las amas de casa fueron menos afectadas en comparación con los restantes 108 casos que tenían diferentes ocupaciones y exposición a los diferentes factores de riesgo considerados en el estudio. Las amas de casa son las menos expuestas a traumas y a factores ambientales.
Entre los factores de riesgo que se siguen estudiando se encuentran la actividad física intensa, los hábitos tóxicos sobre todo el tabaquismo, la comorbilidad por enfermedades crónicas, por enfermedades inflamatorias crónicas autoinmunes, infecciones virales, traumatismos, la edad mayor de 50 años, el predominio en varones fue de 2:1 con respecto a mujeres y en la raza blanca (caucásica). Hubo momentos que se consideró la posibilidad de la infección previa por retrovirus, y entre ellos el VIH, pero estudios posteriores de casos con buen nivel de evidencias lo descartó. En estudios epidemiológicos revisados y de estudio de casos está demostrado, a pesar que se sigue investigando, la influencia de los factores de riesgo que se tuvieron en cuenta en el presente estudio y que se relacionan con menor sobrevida, independientemente de la forma clínica de la enfermedad.20,21,22,23,24,25
La comorbilidad, igualmente, se señala en la literatura que acorta la supervivencia de los pacientes, sobre todo en los mayores de 65 años,1,2,3,5,14,15,21,24,25,26,27,28,29) lo cual es entendible pues a partir de esas edades las comorbilidades se hacen mucho más frecuentes.
El presente estudio sí demuestra que la asociación de otras enfermedades crónicas y antecedentes familiares de la enfermedad influyen a mediano plazo en la supervivencia, independientemente de la forma clínica.
No se demostró diferencias significativas en las curvas de supervivencia de los pacientes que recibieron tratamiento con riluzol y los que no lo recibieron. Ya se ha reportado en diversos estudios internacionales y fuentes bibliográficas; hay defensores y retractores.1,2,3,5,14,15,29,30,31,32,33,34,35,36,37) El riluzol puede tener un efecto mayor en los pacientes más jóvenes y en aquellos con un retraso menor en el diagnóstico. También se ha demostrado que tres tratamientos médicos prolongan la supervivencia: ventilación no invasiva (VNI) cuando es necesaria la asistencia respiratoria, alimentación con gastrostomía cuando hay disfagia y el riluzol.30,31,32,33,34,35,36)
Como un tratamiento farmacológico que ha demostrado prolongar la supervivencia en una enfermedad neurodegenerativa, el riluzol es de gran interés tanto teórico como práctico, pero no se sabe si la mejora de la supervivencia se produce a lo largo del curso de la enfermedad o solo en ciertas etapas, referidas fundamentalmente, al paciente joven, con poco tiempo del diagnóstico y forma clínica espinal.33
Existen complicaciones que son las que producen directamente la muerte del paciente, de ellas la más frecuente es la insuficiencia respiratoria, causada por la propia enfermedad neurológica, neumonías, broncoaspiración y tromboembolismo pulmonar principalmente. Las estimaciones oscilan entre el 65 % y el 89 %.32) Otras son desnutrición, úlceras por presión por el encamamiento e infecciones urinarias complicadas por cateterismo vesical.25,26,27,28,29,30
Finalmente, queremos expresar que no basta con una consulta multidisciplinaria para la atención de estos casos. Internacionalmente se postula que deben existir en alguna institución de salud unidades multidisciplinarias para la atención especializada e individualizada de estos pacientes, que abarcan no solo asistencia ventilatoria en los casos avanzados, sino garantizar una nutrición adecuada y otros cuidados necesarios según la etapa evolutiva.33,34,37
La limitación del estudio estuvo al determinar las causas directas de muerte por no cerrarse adecuadamente los certificados de defunción. Dentro del grupo de las enfermedades neuromusculares existen varias enfermedades, la ELA es una de ellas. Si bien está establecido que el deterioro neurológico conlleva a la muerte, existen complicaciones que son las que producen directamente la muerte del paciente. Hubiera sido muy útil haber conocido las causas fundamentales del deceso para completar este estudio.
La mayoría de los pacientes no tenían análisis genético en las historias clínicas por lo cual no pudo abordarse este aspecto y su análisis.
Conclusiones
En nuestra serie las edades más afectadas estuvieron a partir de los 51 años. En general el mayor número de pacientes murió entre los 2 y 5 años a partir del diagnóstico. La mayor sobrevida estuvo en el grupo de 51 a 60 años, algunos alcanzaron hasta 10 años, en ellos predominó la forma espinal de la enfermedad. Es de señalar que aún viven 37 pacientes. En los pacientes con comorbilidades, antecedentes familiares de la enfermedad y forma bulbar la sobrevida fue menor. La supervivencia al evaluar la efectividad del tratamiento con riluzol con los que no lo consumieron no fue significativa a pesar de recomendaciones internacionales.

