










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Reumatología
versión On-line ISSN 1817-5996
Rev Cuba Reumatol vol.15 no.2 La Habana ago. 2013
ARTÍCULO DE OPINIÓN Y ANÁLISIS
Granulomatosis de Wegener y embarazo. ¿Riesgo perinatal?
Wegener's granulomatosis and pregnancy. ¿Perinatal risk?
Miguel Ángel Serra Valdés
Especialista de 2do. Grado en Medicina Interna. MSc. Profesor Auxiliar. Consultor de los Servicios Ginecobstétricos.
Hospital General Docente Enrique Cabrera.
RESUMEN
La granulomatosis de Wegener es una vasculitis granulomatosa necrotizante que afecta las vías respiratorias superiores, inferiores y los glomérulos. Ésta y la poliangitis microscópica son enfermedades vasculares asociadas con anticuerpos anticitoplasma del neutrófilo. La granulomatosis de Wegener es una enfermedad rara, de causa aún no definida, con incidencia de 0.4 casos por cada 100,000 habitantes. La revisión bibliográfica pone al día los conocimientos acerca de esta enfermedad que permitan al profesional tenerla en mente para diagnosticarla con oportunidad y enfrentar el riesgo perinatal.
Palabras clave: vasculitis granulomatosa necrotizante, glomérulos, anticuerpos anticitoplasma del neutrófilo, ANCA, granulomatosis de Wegener. Riesgo perinatal.
ABSTRACT
Wegener's granulomatosis is a necrotizing granulomatous vasculitis that affects the upper respiratory tract, lower and glomeruli. This and microscopic polyangiitis are vascular diseases associated with neutrophil cytoplasmic antibodies. Wegener's granulomatosis is a rare disease, cause not yet defined, with an incidence of 0.4 cases per 100,000 inhabitants. The literatura review updates the knowledge about this disease that allow the professionals to diagnose keep it in mind to try and it faces the perinatal risk.
Keywords: necrotizing granulomatous vasculitis, glomeruli, neutrophil cytoplasmic antibodies, ANCA, Wagener's granulomatosis, perinatal risk.
INTRODUCCIÓN
La granulomatosis de Wegener (GW) es una vasculitis sistémica, necrosante y granulomatosa, de causa aún desconocida, que afecta el tracto respiratorio superior e inferior, así como el riñón principalmente, aunque cualquier otro órgano puede ser afectado. Heinz Klinger realizó los primeros informes en 1931 y Friederich Wegener en 1939; sin embargo, la descripción final y la denominación como GW la realizaron Godman y Churg en 1954, quienes describieron la tríada característica de la enfermedad.1,2
La GW , el síndrome de Churg-Strauss y la poliangeítis microscópica son vasculitis que afectan vasos pequeños. Por su asociación con los anticuerpos anticitoplasma del neutrófilo (ANCA) son llamadas vasculitis ANCA positivas y de ellas la GW es la más frecuente. Su diagnóstico se basa en las manifestaciones clínicas, las imágenes, la biopsia de los órganos afectados y la presencia de ANCA en el suero.3-8 Sin embargo, inicialmente existen formas limitadas sin afectación multiorgánica donde el diagnóstico se retrasa.4,6,7,8
El objetivo de este artículo es revisar las manifestaciones clínicas, diagnóstico y tratamiento de la GW y especial atención cuando está vinculada al embarazo, pues constituye riesgo perinatal.5,7,8 Dentro del gran capítulo de las vasculitis su conocimiento debe ser tanto del internista, como el reumatólogo, hematólogo, inmunólogo y ginecobstetra. Contribuye el artículo, desde el punto de vista científico-documental, a la competencia de los profesionales de las especialidades mencionadas y al personal en formación desde el punto de vista docente.
DESARROLLO
En 1866, Kussmaul y Maier realizaron la descripción arquetípica de la vasculitis sistémica cuando comunicaron la dolencia de un oficial de sastre de 27 años que tenía lo que hoy en día se conoce como poliarteritis nudosa. Lo describieron como «uno de esos pacientes para los cuales uno puede dar antes el pronóstico que el diagnóstico. La primera impresión era la de un alma perdida, cuyos días estaban contados.5 Durante muchos años, el pronóstico asociado con muchas de las formas de vasculitis sistémica era nefasto del mismo modo.
Cuando se describió por primera vez la forma sistémica de la GW , por ejemplo, era esencialmente un diagnóstico fatal. Los años recientes han visto la confluencia de múltiples eventos que han mejorado drásticamente los resultados de las personas afectadas de vasculitis sistémica.6 En primer lugar, el mayor grado de alerta sobre estas raras enfermedades ha dado lugar a mayores tasas de diagnóstico, lo que permite un tratamiento más temprano y efectivo. En segundo lugar, se han producido avances en el tratamiento, inicialmente mediante la introducción de la ciclofosfamida y otros fármacos citotóxicos para el tratamiento de la vasculitis grave. Posteriormente mediante la evolución de estos tratamientos, diseñados para minimizar la exposición a los mismos, preservar tanto la calidad como la cantidad en años de vida de los pacientes. Finalmente, se han producido avances en las pruebas diagnósticas, incluyendo la identificación de biomarcadores que facilitan el diagnóstico temprano.8-13
GRANULOMATOSIS DE WEGENER COMO VASCULITIS SISTÉMICA
La GW es una enfermedad poco frecuente con una prevalencia estimada en EEUU de 3 casos por 100.000 habitantes, afectando ambos sexos por igual. En general existen muy pocos reportes de casos en la literatura. Los estudios epidemiológicos sobre las vasculitis primarias en Latinoamérica son escasos y los pocos que se han realizado, generalmente son estudios en poblaciones específicas en Estados Unidos y Europa; en algunos de estos estudios se sugiere que existe un incremento de estas enfermedades en la última década. No existen frecuencias estimadas reportadas por otros países. Puede ocurrir a cualquier edad aunque la media se sitúa en los 40 años y predomina en la raza blanca. El 15 % de los pacientes tiene menos de 19 años, y sólo rara vez aparece antes de la adolescencia.3,4,7,8,12,13
ETIOLOGÍA Y PATOGENIA
La etiología es desconocida. No se ha comprobado la existencia de predisposición genética ni asociación con antígenos del complejo mayor de histocompatibilidad. Sin embargo, se acepta que el sistema inmune participa en su patogenia. Se considera que un antígeno extraño inhalado y/o propio localizado en las vías respiratorias constituye el factor que desencadena una respuesta anómala del sistema inmune, con una regulación inadecuada de sus componentes celulares, lo que provoca una reacción inflamatoria tanto local en las vías respiratorias como sistémica. Esta participación patogénica del sistema inmune se manifiesta en la infiltración, centrada en los vasos de los tejidos afectados por polimorfonucleares, macrófagos y linfocitos, de predominio fenotípico T. Asimismo, se sintetizan anticuerpos frente a antígenos presentes en los polimorfonucleares, que producen un patrón de tinción citoplasmática granular en estas células mediante inmunofluorescencia indirecta (ANCA-C). 8-13
Espectro clínico. El espectro clínico es muy variado y esto hace que en ocasiones se retrase el diagnóstico. Al inicio son muy frecuentes los síntomas inespecíficos (artromialgias, fiebre y anorexia), siendo el motivo de consulta más común síntomas de vía respiratoria superior, inferior o ambas. Es muy frecuente que consulten por síntomas pulmonares como tos, hemoptisis, dolor torácico o disnea, encontrándose alteraciones radiológicas tipo nódulos o infiltrados en un 45 %. Más del 65 % consultan por síntomas de sinusitis y más de un tercio por clínica nasal como rinorrea, obstrucción, úlceras o epistaxis. Un 25 % debuta con manifestaciones otológicas, sobre todo otitis media serosa y pérdida de audición. La afectación renal inicialmente es poco frecuente (un 18 %) pero es la causa de muerte en los pacientes no tratados pues tiene peor pronóstico.4
Los síntomas de presentación más frecuentes son los del tracto respiratorio superior en más de 90 % de los casos. Pueden aparecer como manifestación inicial sin afección del riñón y la GW se considera limitada y tiene mejor pronóstico. Los signos y síntomas usuales consisten en descarga nasal purulenta, fiebre, tos, hemoptisis, dolor de los senos paranasales, ulceración de la mucosa paranasal y deformidad de la nariz en silla de montar. La estenosis subglótica y la estenosis del árbol bronquial son menos frecuentes, aunque graves; otros síntomas son disfonía, disnea, estridor o sibilancias mal diagnosticadas como asma, sin embargo, la afección del tracto traqueobronquial puede ser asintomática. La afección pulmonar se manifiesta por nódulos asintomáticos, infiltrado pulmonar y hemoptisis, cuya frecuencia es de 5 a 45 %, con mal pronóstico y mortalidad de 50 %.7-13
El daño renal es un distintivo de la GW ; la glomerulonefritis se presenta en más de 75 % de los casos; no obstante, la insuficiencia renal grave al inicio de la GW se observa de 11 a 17 %. La glomerulonefritis focal y segmentaría es la lesión renal característica con cilindros granulares (55 %) y eritrocitarios (14 %), así como proteinuria mayor a 1 g/día hasta en 60 %. La insuficiencia renal crónica se observa del 11 al 32 % y es una manifestación de mal pronóstico, en especial cuando se presenta como glomerulonefritis rápidamente progresiva.7-13
Las manifestaciones músculo esqueléticas ( 60 a 80 %) se caracterizan por artritis no erosiva, artralgias y mialgias. También se observan en otras vasculitis sistémicas. Las manifestaciones cutáneas se han informado hasta en un 20 % como manifestación inicial y alrededor de 50% durante el curso de la GW. Son inespecíficas; las más frecuentes son la púrpura palpable, vesículas, pápulas, costras, úlceras, entre otras. Cuando aparecen en forma inicial obligan a investigar afección en otros órganos.7-13 Las manifestaciones oculares se presentan en el 20 a 50 % de los casos e incluye desde conjuntivitis hasta inflamación grave como queratitis ( 12 a 20 %), episcleritis y escleritis ( 12 a 27 %), uveítis ( 2 a 7 %) y neuritis óptica ( 12 a 16 %). La dacriocistitis se ha descrito hasta en el 18 %, así como de los tejidos de la órbita. La proptosis es una manifestación distintiva de la GW , se presenta en 15 a 57 %. Estas manifestaciones son secundarias a vasculitis de los vasos de la retina o a extensión del proceso granulomatoso de los senos paranasales o de los tejidos blandos hacia la órbita. La afección ocular es una manifestación primordial de la GW que la distingue de otras vasculitis sistémicas y de otras vasculitis ANCA positivas.7-13
La afección neurológica inicial se encuentra en menos de 5 % y durante el curso de la enfermedad en 22 a 54 %. La actividad a nivel del sistema nervioso central (10 %) se caracteriza por infartos cerebrales, hemorragia, inflamación cerebral, meningitis crónica, déficit focal o crisis convulsivas, diabetes insípida o cefaleas crónicas. Las alteraciones del sistema nervioso periférico (22 %) son la mononeuritis múltiple y la polineuritis. Estas manifestaciones neurológicas, excepto la diabetes insípida también, se pueden observar en otras vasculitis ANCA positivas. Las afecciones a nivel cardiovascular se observan en 15 %, las más frecuentes son pericarditis, insuficiencia cardiaca, miocarditis y vasculitis coronaria y no son específicas de esta vasculitis. A nivel digestivo las manifestaciones ( 4 a 10 %) más frecuentes son dolor abdominal, diarrea, hemorragia y perforación intestinal. En estudios post mortem, la afección gastrointestinal histopatológica se ha confirmado hasta en un 39 %.7-13
DATOS DE LABORATORIO
La sangre muestra anemia, trombocitosis o leucocitosis en 30 a 40 % de los pacientes. La hipergammaglobulinemia policlonal ocurre en 50 %. Los niveles del complemento generalmente son normales. La velocidad de sedimentación globular y la proteína C reactiva están aumentadas durante la actividad de la GW ; el examen general de orina muestra sedimento urinario activo (leucocituria o cilindros leucocitarios, eritrocitrocitos principalmente dismórficos o cilindros hemáticos o granulosos). Estos resultados son inespecíficos y se pueden encontrar en otras vasculitis. Los ANCA son autoanticuerpos dirigidos contra constituyentes de los gránulos de los neutrófilos con una alta especificidad (98 %). Los de patrón citoplasmático (ANCA-c) con especificidad PR 3 son de utilidad en el diagnóstico inicial y en el monitoreo de la respuesta terapéutica. Los niveles de ANCA-c elevados ( 90 a 95 %) se encuentran en la GW generalizada y en la enfermedad localizada en 40 a 70 %. Las variaciones en los niveles de los ANCA-c generalmente se correlacionan con la actividad; no obstante, sus títulos pueden persistir hasta en 30 a 40 % de los casos en remisión clínica. Así mismo, su incremento no siempre indica recaída.7-13
ESTUDIOS DE IMAGEN

En las radiografías simples y tomografías computarizadas de los senos paranasales se puede corroborar el diagnóstico de sinusitis. Figuras 1

En las radiografías de tórax se observan nódulos únicos o múltiples de diferentes tamaños en los lóbulos inferiores; Figura 2 las cavitaciones también son frecuentes. Estas alteraciones radiológicas son características de la GW y se consideran en los criterios de clasificación del American College of Rheumatology, aunque se puede observar que los nódulos son no fijos en otras vasculitis como el síndrome de Churg-Strauss, la artritis reumatoide, entre otros.
Las opacidades alveolares difusas se deben a hemorragia pulmonar, atelectasias por estenosis bronquial, infiltrados uni o bilateral que frecuentemente se diagnostican como neumonía y derrame pleural.
La tomografía computarizada de alta resolución puede hacer evidente información no observada en la radiografía de tórax: estenosis traqueobronquial, engrosamientos de la pared de los bronquios, nódulos fijos, infiltrado reticular, entre otros. 32 Los nódulos pulmonares se encuentran dentro de los criterios de clasificación del American College of Rheumatology.7-13
ESTUDIOS HISTOPATOLÓGICOS
La GW se caracteriza por granulomas necrosantes del tracto respiratorio superior e inferior y vasculitis necrosante o granulomatosa de vasos pequeños, generalmente en el pulmón y glomerulonefritis focal y segmentaria.

En el pulmón la necrosis tisular se puede encontrar en ausencia de vasculitis. El patrón clásico histológico es la necrosis geográfica (zonas grandes e irregulares que sustituyen el parénquima). La necrosis varía desde la semejante a la caseosa a la de aspecto sucio basofílico o supurativo, con neutrófilos necróticos y otras células inflamatorias. [Figura 3]
Entre el proceso inflamatorio se puede observar histiocitos dispuestos en palizada en la periferia de una zona de necrosis. La vasculitis en arterias y venas puede ser granulomatosa, linfocítica o histiocítica.10,14

En el tejido renal, la lesión histológica más común es una glomerulonefritis necrosante con semilunas. Las lesiones agudas varían desde una glomerulonefritis focal y segmentaria con necrosis fibrinoide con semilunas, hasta una glomerulonefritis difusa necrosante y semilunas que afecta casi todos los glomérulos. [Figura 4]

En las lesiones agudas se observa necrosis fibrinoide, infiltrado de neutrófilos leve y cariorexis; los capilares glomerulares adyacentes a estos segmentos pueden contener trombos. [Figura 5] También puede existir hipercelularidad endocapilar y paredes capilares engrosadas.10,14
DIAGNÓSTICO
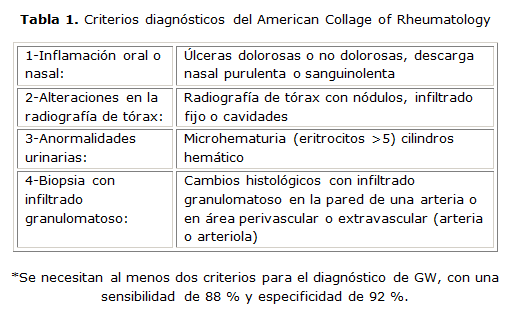
Es necesario distinguir esta entidad de otras vasculitis, del síndrome de Goodpasture, de los tumores de las vías respiratorias superiores y del pulmón, el granuloma de la línea media, de ciertas enfermedades infecciosas como la histoplasmosis, las leishmaniosis mucocutáneas, el rinoescleroma, así como de otras enfermedades granulomatosas no infecciosas. También debe distinguirse de la granulomatosis linfomatoide, que consiste en la proliferación de células B positivas para virus de Epstein-Barr que conlleva una reacción exhuberante de células T. La granulomatosis linfomatoide se caracteriza por lesiones pulmonares, cutáneas, del sistema nervioso central y renal donde un grupo de células linfocitoides y plasmacitoides atípicas infiltran al tejido no linfoide en forma angioinvasora.6-14 [Tabla 1]
TRATAMIENTO15-34
Las vasculitis ANCA positivas son un grupo de vasculitis de pequeño vaso que incluyen la granulomatosis de Wegener, la poliangeítis microscópica, el síndrome de Churg-Strauss y las vasculitis limitadas al riñón. En los últimos años el pronóstico vital de estas entidades ha mejorado mucho, pero sigue existiendo una alta mortalidad entre las personas de edad avanzada y los pacientes que inician el cuadro con una insuficiencia renal avanzada.
El esquema de tratamiento que ha permitido dar este salto adelante en la evolución de las vasculitis ANCA positivas se describe a continuación. Para las formas localizadas, puede ser adecuado el tratamiento con cotrimoxazol asociado o no a prednisona y para las formas llamadas early systemic el tratamiento de elección se considera el metotrexato junto con los corticoides. En las formas sistémicas con afectación renal y valores de creatinina <500 mmol/l, la mejor opción terapéutica son los bolos de ciclofosfamida y corticoides para la inducción de la remisión, y azatioprina y corticoides para el mantenimiento de ésta al menos hasta los 2 años del diagnóstico.
Para las formas más severas con insuficiencia renal y valores de creatinina >500 mmol/l, la plasmaféresis asociada a ciclofosfamida oral y corticoides se considera la pauta más adecuada, superando la plasmaféresis a los bolos de metilprednisolona en cuanto a supervivencia renal a un año de seguimiento. Para el mantenimiento de la remisión, la pauta más validada es la que incluye azatioprina y corticoides. Uno de los problemas más importantes por resolver es la alta tasa de efectos secundarios observados con estas pautas inmunodepresoras, así como el momento de parar la inmunodepresión.
Como alternativas a la pauta propuesta, especialmente en casos de efectos secundarios o intolerancia a la ciclofosfamida o azatioprina, se dispone de micofenolato mofetil. Están en marcha distintos ensayos clínicos que comparan la pauta clásica con este fármaco. Finalmente, en aproximadamente un 10 % de casos, el tratamiento anteriormente descrito no es efectivo y se precisa rescate. Existe poca evidencia sobre la mejor opción en estos casos, pero parece que rituximab puede ser una buena opción.
WEGENER Y EMBARAZO
Los temas relacionados con la fertilidad se han convertido en un nuevo reto para el tratamiento de las pacientes con vasculitis sistémica primaria. A diferencia de muchas de las formas de autoinmunidad (como la enfermedad tiroidea, la artritis reumatoide o el lupus eritematoso sistémico), la vasculitis sistémica primaria no afecta a las mujeres de manera preferencial. Dado que estas enfermedades son infrecuentes, nuestro conocimiento de cómo se ve afectado el embarazo por la vasculitis es limitado, y procede principalmente de comunicaciones de muy escasos casos y de series muy limitadas.
El diagnóstico y la evaluación de la vasculitis durante el embarazo puede ser muy difícil. Los reactantes de fase aguda pueden ser un marcador no fiable de inflamación durante el embarazo. Por ejemplo, la velocidad de sedimentación globular suele utilizarse para monitorizar a las pacientes con vasculitis sistémica. Sin embargo, el embarazo se asocia con una elevación del fibrinógeno sérico, lo que da lugar a una elevación de la velocidad de sedimentación. También pueden verse durante el embarazo elevaciones de la proteína C reactiva, independientemente de la existencia de un brote de vasculitis.5
La planificación preconcepcional es particularmente crucial para las pacientes con vasculitis sistémica. Debe considerarse cuidadosamente, no sólo el estado general de salud de la paciente, sino también el estado de la vasculitis subyacente y los fármacos requeridos para mantener la remisión. Idealmente, el embarazo debería producirse sólo después de una remisión prolongada. El embarazo debe ser tratado por un equipo multidisciplinario, incluyendo un reumatólogo y un obstetra de alto riesgo, y la madre debe someterse a frecuente monitorización en busca de evidencia de actividad de la enfermedad. Las enfermedades que tienen una patogenia autoinmune y afectación sistémica como la que nos ocupa se encuentran enmarcadas dentro del riesgo perinatal, si tenemos en cuenta también la afectación de órganos fundamentales como pulmones, riñones, corazón y otros. La comorbilidad se suma, más los efectos de la terapéutica sobre la madre y el feto.5,35,36
En cualquier momento del embarazo pueden presentarse nuevos diagnósticos de granulomatosis de Wegener; las comunicaciones de casos describen el inicio de la enfermedad durante el primer trimestre, el segundo el tercero y en el posparto inmediato. Existen varias descripciones de brotes presentándose inmediatamente antes del aborto espontáneo o voluntario suscitando la posibilidad de que el parto ocasione cambios inmunológicos que predispongan al brote de la enfermedad. Sin embargo, el final del embarazo no ocasiona la remisión en todos los casos. Aunque la mayoría de las descripciones de casos hacen referencia a mujeres con enfermedad limitada, también existen algunas publicaciones de embarazadas con formas de granulomatosis de Wegener potencialmente mortales.35,36 Cuando se evalúa a una paciente con una vasculitis sistémica conocida, respecto a la posibilidad de un brote, son de utilidad algunas recomendaciones. En primer lugar, la infección puede simular muchas de las manifestaciones de vasculitis. Por ello es importante descartar la posibilidad de focos sépticos antes y durante la administración de tratamientos inmunodepresores, especialmente cuando la paciente no responde como se espera. En segundo lugar, independientemente de la forma de vasculitis, la historia de la paciente no resulta necesariamente predictiva de los futuros eventos. Una paciente que tiene un brote de la enfermedad durante un embarazo puede, o no, tener un brote durante el siguiente.5
La ciclofosfamida está contraindicada durante el primer y segundo trimestres del embarazo, pero puede considerarse durante el tercer trimestre para el tratamiento de la enfermedad grave. Se ha argumentado que la inmunosupresión agresiva puede ser menos tóxica para el feto que un brote de vasculitis insuficientemente tratado. En algunos casos, la inmunoglobulina por vía intravenosa puede ser una alternativa efectiva cuando la ciclofosfamida no puede utilizarse con seguridad. El metotrexato, que se utiliza con frecuencia para el tratamiento de muchas formas de vasculitis sistémica, se asocia con mal formaciones craneales y de las extremidades cuando se produce la exposición in útero, y por ello está contraindicado durante el embarazo. Puesto que los datos son limitados, sigue desconociéndose la seguridad de un gran número de medicaciones inmunodepresoras durante el embarazo, particularmente de los agentes biológicos. Aunque otros agentes biológicos, como el rituximab, pueden ser seguros durante el embarazo, la experiencia es escasa, por lo que debería evitarse su utilización durante el embarazo siempre que sea posible.5,35,36
No obstante, esto está controvertido, dado que muchos investigadores destacan que la enfermedad durante el embarazo puede ser particularmente difícil de controlar. La granulomatosis de Wegener no tiene un impacto predecible sobre el resultado del embarazo; pero la preeclampsia, la prematuridad, el aborto recurrente, el desprendimiento placentario, la susceptibilidad a las infecciones graves por los inmunosupresores no son complicaciones infrecuentes.37
En resumen, hasta hace poco tiempo, el pronóstico de muchas de las formas de vasculitis era sombrío. Los avances en este campo han permitido empezar a centrarnos en temas relacionados con la calidad de vida, como la fertilidad y la concepción, así como el embarazo en las mujeres afectadas de vasculitis. Dado que las vasculitis sistémicas representan diagnósticos infrecuentes, quedan sin respuesta muchas cuestiones de importancia. Globalmente, parece que las mujeres con vasculitis inactiva puede que no presenten riesgo para las complicaciones durante el embarazo, pero nuestro conocimiento de la interacción entre el embarazo y las formas específicas de vasculitis debe enriquecerse más.
La problemática de los efectos de los medicamentos que consumen sí pueden constituir riesgo. Hay que individualizar los casos. La enfermedad durante el embarazo puede ser particularmente difícil de controlar y la preeclampsia y la prematuridad no son complicaciones infrecuentes.
REFERENCIAS BIBLIOGRÁFICAS
1. Vera-Lastra OL, Halabe-Cherem J, Jara LJ. Vasculitis sistémicas. Conceptos generales. En: Vera-Lastra OL, Halabe-Cherem J, eds. Vasculitis. México DF: Ed. Alfil; 2006. p. 1-4
2. Goodman GC, Churg J. Wegener's granulomatosis: pathology and review of the literature. Arch Pathol. 1954;58:533-53.
3. Osaki S. ANCA-associated vasculitis: diagnostic and therapeutic strategy. Allergol Int. 2007;56:87-96.
4. García Arpa M, Porras Leal L, Romero Aguilera G, Vera Iglesias E, García Rojo M, Sánchez Caminero P, Cortina de la Calle P. Encías en fresa como manifestación inicial de enfermedad de Wegener. Med Cutan Iber Lat Am. 2009;37(5):217-20.
5. Philip Seo, MD, MHS. Embarazo y vasculitis. Rheum Dis Clin N Am. 2007;33:299-317.
6. Wiik AS, Gordon TP, Kavanaugh AF. Cutting edge diagnostic in rheumatology: the rol of patients, clinicians and laboratory scientists in optimizing the use of autoinmune serology. Arthritis Rheum. 2004;51:291-8.
7. Keyla C, Ibañez S, Barba D. Granulomatosis de Wegener. Reporte de un caso. Rev méd cient. 2010;23(2):61-71.
8. Haris M, Koulaouzidis A, Yasir M, Clark S, Kaleem M, Mallya R, et al. Wegener´s Granulomatosis. CMAJ. 2008;178(1):25-6.
9. Vera-Lastra OL. Abordaje diagnóstico de las vasculitis. En: Vera-Lastra OL, Halabe Cherem J, eds. Vasculitis. México DF: Ed. Alfil; 2006. p. 77-99.
10. Flores-Suárez F. Granulomatosis de Wegener. En: Vera-Lastra O, Halabe-Cherem J, eds. Vasculitis. México DF: Ed. Alfil; 2006. p. 179-87.
11. Granulomatosis de Wegener. En Manual A MIR de Reumatología. Colectivo de autores. [documento en Internet]. España. 2008 [citado el 6 de julio de 2013]. 3ed. Academia AMIR. Madrid: 20-21 Disponible en: www.academiamir.com
12. Vera Lastra O, Olvera Acevedo A, McDonal Vera A, Pacheco Ruelas M, Gayosso Rivera JÁ. Granulomatosis de Wegener, abordaje diagnóstico y terapêutico. Gac Méd Méx. 2009;146(2):121-9
13. Salazar, Kenneth. Granulomatosis de Wegener y vasculitis asociadas con anticuerpos anticitoplasma de neutrófilos (ANCA). Revista médica de Costa Rica y Centroamérica. 2008;583:175-7.
14. Schoe F. Vasos sanguíneos: granulomatodis dce Wegener. En Robins y Cotran. Patología estructural y funcional. 7ed. Madrid: Ed. Kumar-Abbas-Fausto editors; 2007. p. 547
15. Pesci A, Manganelli P. Involvement of the respiratory system an ANCA associated systemic vasculitides: clinical and pathologic hallmarks and treatment. Drugs 2007; 8:25-42.Pagnoux C, Teixeira L. Wegener's granulomatosis. Presse Med. 2007;36(5Pt2):860-74.
16.Colectivo de Autores. HARRISON ONLINE 17 ed. > Parte XIII. [libro en Internet]. Trastornos del sistema inmunitario, el tejido conectivo y las articulaciones > Sección 2. Trastornos mediados por mecanismos inmunitarios > Capítulo 306. Síndromes de las vasculitis > Granulomatosis de Wegener [citado el 5 de julio de 2013]. Disponible en: http://www.harrisonmedicina.com/popup.aspx?aID=95157&print=yes
17. Patricia M Fortin, Aaron M Tejani, Ken Bassett, Vijaya M Musini. Inmunoglobulina intravenosa como tratamiento adyuvante para la granulomatosis de Wegener (Revision Cochrane traducida).[documento en Internet]. Biblioteca Cochrane Plus 2009 [citado el 5 de julio de 2013]. Número 3. Oxford: Update Software Ltd. Disponible en: http://www.update-software.com
18. Iglesias-Gamarra, Antonio; Coral, Paola. Historia de las vasculitis primarias en Latinoamérica. Revista colombiana de Reumatología. 2007;14:261-86.
19. Leavitt RY, Fauci AS. The American College of Rheumatology 1990 criteria for the classification of Wegener's granulomatosis. Arthritis Rheum. 1990;8:1101-07
20. Jayne, David; Gaskin, Gill. Randomized trial of plasma exchange or high dosage methylprednisolone as adjunctive therapy for severe renal vasculitis. J Am Soc Nephrol. 2007;18:2180-88.
21. Hiemstra, Thomas, et al. Mycophenolate Mofetil vs Azathioprine for Remission Maintenance in Antineutrophil Cytoplasmic Antibody-Associated Vasculitis. JAMA. 2010;304:2381-88.
22. Calzada Algrávez JL, Jaramillo Ramírez H, Delgadillo Márquez G, Macías Díaz DM. Granulomatosis de Wegener: presentación de un caso clínico y revisión de la bibliografía. Med Int Mex. 2012;28(5):504-07
23. Booth A, Harper L, Hammad T, Bacon P, Griffith M, Levy J. Prospective study of TNFalpha blockade with infliximab in anti-neutrophil cytoplasmic antibody-associated systemic vasculitis. Journal of the American Society of Nephrology 2004; 15(3): 717-21.
24. Chung SA, Seo P. Advances in the use of biologic agents for the treatment of systemic vasculitis. Current Opinion in Rheumatology. 2009;21:3-9.
25. De Groot K, Rasmussen N, Bacon PA, Tervaert JW, Feighery C, Gregorini G, et al. Randomized trial of cyclophosphamide versus methotrexate for induction of remission in early systemic antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis & Rheumatism. 2005;52(8):2461-9.
26. Hellmich B, Lamprecht P, Gross WL. Advances in the therapy of Wegener's granulomatosis.. Current Opinion in Rheumatology 2006; 18(1): 25-32.
27. Jayne D, Rasmussen N, Andrassy K, Bacon P, Tervaert JW, Dadoniene J, et al. A randomized trial of maintenance therapy for vasculitis associated with antineutrophil cytoplasmic autoantibodies. New England Journal of Medicine. 2003;349(1):36-44.
28. The Wegener's Granulomatosis Etanercept Trial (WGET) Research Group. Etanercept plus standard therapy for Wegener's granulomatosis.. New England Journal of Medicine 2005; 352(4): 351-61.
29. Wung PK, Stone JH. Therapeutics of Wegener's granulomatosis. Nature Clinical Practice Rheumatology. 2006;2(4):192-200.
30. Ara J. Tratamiento de las vasculitis ANCA positivas. nefroPLUS. 2010;3(1):28-38.
31. Roccatello D, Baldovino S, Alpa M, Rossi D, Napoli F, Naretto C. Effects of anti-CD20 monoclonal antibody as a rescue treatment for ANCA-associated idiopathic systemic vasculitis with or without overt renal involvement. Clin Exp Rheumatol. 2008;26 Suppl 49:S67-71.
32. Jayne DR, Gaskin G, Rasmussen N, Abramowicz D, Ferrario F, Guillevin L; European Vasculitis Study Group. Randomized trial of plasma exchange or high-dosage methylprednisolone as adjunctive therapy for severe renal vasculitis. J Am Soc Nephrol. 2007;18:2180-8.
33. De Groot K, Harper L, Jayne DR, Flores Suarez LF, Gregorini G, Gross WL, et al.; EUVAS (European Vasculitis Study Group). Pulse versus daily oral cyclophosphamide for induction of remission in antineutrophil cytoplasmic antibody-associated vasculitis: a randomized trial. Ann Intern Med. 2009;150:670-80.
34. Hu W, Liu C, Xie H, Chen H, Liu Z, Li L. Mycophenolate mofetil versus cyclophosphamide for inducing remission of ANCA vasculitis with moderate renal involvement. Nephrol Dial Transplant. 2008;23:1307-12.
35. Besáis N, Moulakakis KG, Lioupis C. Wegener's granulomatosis presenting during pregnancy with acute limb ischemia. J Vasc Surg 2005;42:800-4
36. Luisiri P, Lance NJ, Curran JJ. Wegener's granulomatosis in pregnancy. Arthritis Rheum. 1997;40:1354-60
37. Auzary C, Huong DT, Wechsler B. Pregnancy in patients with Wegener's granulomatosis: report of five cases. Ann Rheum Desease. 2000;59:800-4.
El autor refiere no tener conflicto de intereses.
Recibido: 15 de julio de 2013
Aprobado: 18 de agosto de 2013
Autor para correspondencia: Dr. Miguel Ángel Serra Valdés . E-mail: maserra@infomed.sld.cu
Hospital General Docente Enrique Cabrera. Calzada de Aldabó y Calle E. Altahabana. Municipio Boyeros. La Habana.

