Serviços customizados
Serviços customizados
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
Introducción
Las plaquetas contribuyen a la hemostasia, y la afección de sus procesos bioquímicos heredada o adquirida causa alteraciones de la función plaquetaria. Estos trastornos de función se han asociado a trastornos del tejido conectivo como el síndrome Ehlers-Danlos (SED), síndrome de Marfán, osteogénesis imperfecta, y síndrome del X frágil, aunque no están limitados a estos.1,2
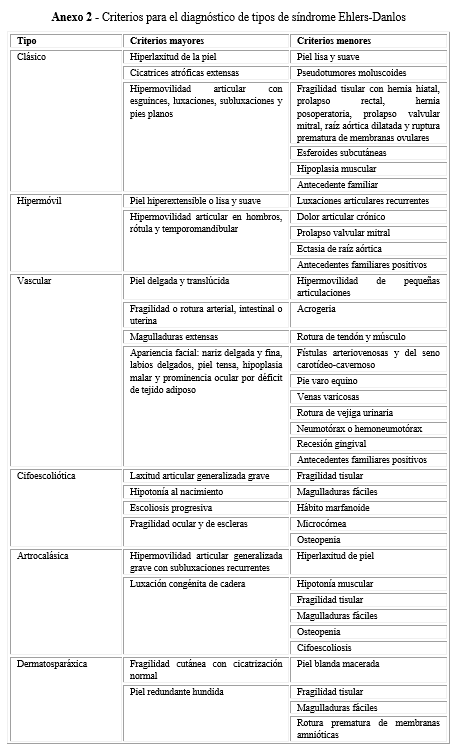
El diagnóstico diferencial entre estos síndromes genéticos puede hacerse clínicamente, pero el espectro de hipermovilidad articular (HMA) y el Ehlers-Danlos en su variante hipermóvil, son defectos hereditarios de tejido conectivo sobrelapados, que se han considerado la misma entidad nosológica con prevalencia estimada entre 1:5000-15 000.3
Clínicamente las fronteras entre la HMA y el SED no están definidas ni por los estudios moleculares, ya que pueden considerarse variantes alélicas con diferente expresión clínica. La HMA tiene una prevalencia estimada del 30 % en diferentes poblaciones y se diagnostica por los criterios de Beighton. El SED tiene entre sus signos clínicos la HMA y los criterios clínicos para diagnóstico han sido reevaluados. Estos criterios no son excluyentes, por lo cual no existen reglas de oro para el diagnóstico clínico diferencial entre estas dos entidades clínicas que tienen en común la afectación en las fibras colágenas.3,4,5,6,7,8,9,10,11
En estos síndromes genéticos las manifestaciones hemorrágicas se han atribuido a fragilidad de tejidos y capilares. También se ha notificado el incremento del tiempo de sangramiento y raras veces trombocitosis o diátesis plasmáticas. Algunos autores han asociado los trastornos en la función o agregación plaquetaria a variantes de Ehlers-Danlos.12,13,14,15,16,17
Los defectos de la función plaquetaria comprenden un grupo heterogéneo de trastornos de la coagulación que varían en la gravedad. Los defectos de liberación se caracterizan por retraso al liberar los gránulos luego de la activación plaquetaria y son causados por anomalías en la transducción de señales de membrana, vías metabólicas internas anormales y mecanismos o estructuras anormales involucradas en las reacciones de liberación. Se estima que el 15,5 % de los estudios de función plaquetaria positivos se relacionan con personas con defectos de la fibra colágena.1,18,19,20,21
El colágeno activa las plaquetas mediante la glicoproteína VI en la vía del inmunorreceptor tirosina activado. El ADP induce la activación de las proteínas receptoras de pareja P2Y12 y P2Y1 G. La epinefrina activa el receptor α2A-adrenérgico y todas estas activaciones confluyen en la vía de la integrina. Entre los afroamericanos hay variantes genómicas que se relacionan con la expresión de PEAR 1 dosis dependiente.18,19,20
Una variante intergénica media en la reacción con ADP en un loci del cromosoma 18q 22.3 que contiene una caja de genes FBXO15 y que interactúa en esta cascada de activación plaquetaria. Específicamente en los afroamericanos, se reportan asociaciones con trombopatías.18,19,20
La zona periférica de las plaquetas participa en la adherencia, agrupación plaquetaria, liberación de gránulos y también están los fosfolípidos plaquetarios, que constituyen la superficie donde reaccionan las proteínas de la coagulación y enzimas plaquetarias promotoras de la agregación como el tromboxano A2. Los elementos de matriz extracelular comprenden entre el 30-50 % de la proteína total de la plaqueta. El colágeno estimula la fosforilación de la cadena ligera de la miosina II y mantiene el citoesqueleto plaquetario y la estabilidad del trombo.1,20,21,22
Teniendo en cuenta que en la HMA y el SED se presentan defectos en la síntesis o activación del colágeno u otras proteínas relacionadas con la matriz extracelular, el estudio de agregación plaquetaria puede mostrar afectaciones cualitativas relacionadas con la función de estas células que constituyan un endofenotipo útil para el diagnóstico diferencial de estas dos entidades clínicas.
Métodos
Se revisaron 353 historias clínicas de pacientes con diagnóstico de HMA o SED en cualquiera de sus variantes, atendidos en el servicio de Genética Clínica del Hospital Pediátrico William Soler desde septiembre del 2009 a septiembre del 2012, en las edades comprendidas entre los 5 y 18 años.
Los pacientes fueron evaluados por medio del interrogatorio y examen físico, y se remitieron a oftalmología, ecocardiografía, ortopedia y fisiatría, como corresponde en la aplicación del método clínico. Los observadores fueron 4 genetistas clínicos que aplicaron los criterios de Beighton para el diagnóstico de HMA y un criterio mayor y uno o dos menores para cada tipo de SED, según Villefranche (1997).3,4,8,9
Se incluyeron solo los pacientes que tenían diagnóstico clínico sin ambigüedades, según los criterios referidos, que fueran mayores de 5 años con resultados completos del estudio de agregación plaquetaria documentados en su historia clínica. En estos casos, se indicó el estudio luego de la identificación de los signos clínicos y no por la sintomatología relativa a trastornos de coagulación.
Los estudios de agregación plaquetaria fueron realizados por dos especialistas del Instituto de Hematología en el mismo laboratorio. Luego de 15 días sin exposición a medicamentos en estos pacientes y con ayuno de 8 h se colectaron las muestras por venipuntura con citrato de sodio al 3,2 % y se procesaron inmediatamente.
Los estudios se realizaron en el agregómetro PAP 8, BIO/DATA Corporation y el coagulómetro Star-8, Otago; las muestras se centrifugaron en la centrífuga PDQ Tm BIO/DATA Corporation y se utilizaron los agonistas difosfato de adenosina (ADP), epinefrina y colágeno, BIO/DATA Corporation.
Principio de la agregometría
En un tubo de vidrio se agita el plasma rico en plaquetas (PRP). La adición de un agonista activa las plaquetas que se agregan en presencia de fibrinógeno. La agregación se mide por un principio turbidimétrico descrito por Born, que se basa en la diferencia en la transmisión de la luz en el PRP comparándolo con el plasma pobre en plaquetas (PPP).2
Principio de la disponibilidad de los fosfolípidos plaquetarios
Cuando las plaquetas son estimuladas por bajas concentraciones de trombina o colágeno, ocurre un movimiento transmembrana de los fosfolípidos procoagulantes (flip-flop) que resulta en un incremento de la fosfatidilserina y del fosfatidilinositol en la región externa de la membrana. Son estos últimos fosfolípidos los que proveen la superficie catalítica para la activación del factor X y de la protrombina. La capacidad de las plaquetas estimuladas de facilitar la activación de la protrombina y del factor X es lo que se denominaba factor 3 plaquetario. Por ello es que se habla de la disponibilidad de los fosfolípidos plaquetarios; este se hace disponible por acción de los agonistas que inducen la agregación plaquetaria y también por partículas de carga eléctrica negativa, como el celite y el caolín.2
Los resultados de estos pacientes fueron procesados en este estudio aplicando métodos de la estadística descriptiva como porcentajes y pruebas de hipótesis para demostrar las diferencias entre los grupos, con nivel de significación estadística de 0,05.
Resultados
Se encontraron trastornos de agregación plaquetaria en 79 de 86 pacientes (92 %): De los 65 casos con HMA, solo en 7 el estudio fue negativo (10 %) y los 21 con diagnóstico de SED tuvieron el estudio positivo. De estos casos con diagnóstico inicial de HMA y estudio negativo, en su evolución 2 de ellos fueron diagnosticados con síndrome de Williams y otro con síndrome MASS, entidades en las que no se afecta la fibra colágena sino la elastina y fibrilina 1, respectivamente. Si excluimos estos casos, solo sería negativo el estudio en un 6 % de la muestra (5 casos de 84) y positivo en el 94 % (Tabla 1).
Tabla 1 Resultados de estudios de agregación plaquetaria en pacientes con hipermovilidad articular y síndrome Ehlers-Danlos
| Entidad | Normal | Alterado | Total |
|---|---|---|---|
| Hipermovilidad articular | 7 | 58 | 65 |
| Síndrome Ehlers-Danlos | 0 | 21 | 21 |
| Total | 7 | 79 | 86 |
Fuente: Historias clínicas del Servicio de Genética del Hospital Pediátrico William Soler.
En cuanto al tipo de defecto por separado fue más frecuente la alteración de la agregación con ADP en la HMA, mientras que en el SED con la disponibilidad de los fosfolípidos plaquetarios (Tabla 2).
Tabla 2 Tipo de defecto de la agregación plaquetaria según diagnóstico de hipermovilidad articular y síndrome Ehlers-Danlos
| Entidad | ADP No. % | Epinefrina No. % | Colágeno No. % | Fosfolípidos No. % | Total | ||||
|---|---|---|---|---|---|---|---|---|---|
| Hipermovilidad articular | 43 | 66 | 28 | 43 | 10 | 15 | 11 | 16 | 65 |
| Síndrome Ehlers-Danlos | 12 | 57 | 10 | 47 | 8 | 38 | 14 | 66 | 21 |
| Total | 55 | 63 | 38 | 44 | 18 | 20 | 25 | 29 | 86 |
ADP: difosfato de adenosina.
Fuente: Historias clínicas del Servicio de Genética del Hospital Pediátrico William Soler
Estos defectos de agregación se presentaron aislados en 24 casos con HMA (37 %) y 9 con SED (43 %). La combinación de respuesta a diferentes agonistas fue más frecuente en ambos grupos de pacientes (Tabla 3). Las respuestas más representativas en la HMA estuvieron relacionadas con el ADP y en el SED con la disponibilidad de los fosfolípidos plaquetarios.
Tabla 3 Defecto de agregación plaquetaria aislado o combinado en pacientes con hipermovilidad articular y síndrome Ehlers-Danlos
| Defecto | Hipermovilidad articular No. % | Ehlers-Danlos No. % | Total | |||
|---|---|---|---|---|---|---|
| Aislado | 24 | 37 | 9 | 43 | 33 | 38 |
| Combinado | 41 | 63 | 12 | 57 | 53 | 62 |
| Total | 65 | 21 | 86 | |||
Fuente: Historias clínicas del Servicio de Genética del Hospital Pediátrico William Soler.
La asociación de varios defectos entre sí fue más representativa con la combinación ADP con epinefrina en 24 pacientes con HMA (37 %), y para el SED en 7 casos (33 %) la afectación de todos los parámetros estudiados ADP-epinefrina-colágeno-fosfolípidos, asociación que solo se presentó en un caso con HMA (Tabla 4).
No hubo pacientes con las combinaciones epinefrina-fosfolípidos; colágeno-fosfolípidos; ADP-colágeno-fosfolípidos y epinefrina-colágeno-fosfolípidos. Si sumamos los casos que tuvieron afectación aislada o combinada que involucrara los fosfolípidos plaquetarios en la muestra de HMA serían 14/65 (18 %) y los SED 11/21 (52 %).
Tabla 4 Defectos en la agregación plaquetaria por tipos según diagnóstico de hipermovilidad articular o síndrome Ehlers-Danlos
| Agonistas | Hipermovilidad No. % | Ehlers-Danlos No. % | Total | ||
|---|---|---|---|---|---|
| ADP | 16 | 24,6 | 3 | 14 | 19 |
| Epinefrina | 0 | - | 1 | 4,7 | 1 |
| Colágeno | 3 | 4,6 | 1 | 4,7 | 4 |
| Fosfolípidos | 5 | 7,6 | 4 | 19 | 9 |
| ADP-epinefrina | 24 | 37 | 1 | 4,7 | 25 |
| ADP-colágeno | 1 | 1,5 | 0 | 1 | |
| ADP-fosfolípidos | 3 | 4,6 | 3 | 14 | 6 |
| Epinefrina-colágeno | 1 | 1,5 | 0 | - | 1 |
| Colágeno-fosfolípidos | 0 | - | 0 | - | |
| Epinefrina-fosfolípidos | 0 | - | 0 | - | |
| ADP-epinefrina-colágeno | 8 | 12 | 0 | - | 8 |
| ADP-epinefrina-fosfolípidos | 3 | 4,6 | 1 | 4,7 | 4 |
| ADP-colágeno-fosfolípidos | 0 | - | 0 | - | - |
| Epinefrina-colágeno-fosfolípidos | 0 | - | 0 | - | - |
| ADP-epinefrina-colágeno-fosfolípidos | 1 | 1,5 | 7 | 33 | 8 |
| Fosfolípidos como factor común | 12 | 16,46 | 11 | 52,38 | 23 |
| Total | 65 | - | 21 | - | 86 |
ADP: difosfato de adenosina.
Fuente: Historias clínicas del servicio de genética del Hospital Pediátrico William Soler.
Los 3 casos con Ehlers-Danlos en su forma vascular tuvieron combinación de trastornos de agregación con ADP-epinefrina, colágeno y fosfolípidos; 3 con la variante hipermóvil se asociaron a ADP-fosfolípido y los únicos 3 pacientes con la forma clásica variante mitis solo mostraron disminución con ADP.
La respuesta aislada a agonistas como la epinefrina tuvo diferencia estadísticamente significativa (p=0,03), así como la combinación de respuesta a ADP-epinefrina (p=0,0002) y ADP-epinefrina-colágeno (p=0,048), que estuvieron relacionadas con el diagnóstico de HMA. Así los trastornos de la liberación de gránulos pueden asociarse a este diagnóstico clínico (Tabla 5).
Tabla 5 Comparación de combinaciones de agonistas en resultados de estudios de agregación plaquetaria de pacientes con hipermovilidad articular o síndrome Ehlers-Danlos
| Variables | Hipermovilidad articular N=65 | Ehlers-Danlos N=21 | Z | p |
|---|---|---|---|---|
| ADP | 0,24 | 0,14 | 0,96 | 0,16 |
| Epinefrina | 0,00 | 0,047 | -1,75 | 0,03 |
| Colágeno | 0,046 | 0,047 | 0,019 | 0,49 |
| Fosfolípidos | 0,076 | 0,19 | -1,48 | 0,06 |
| ADP-epinefrina | 0,37 | 0,047 | 2,83 | 0,0002 |
| ADP-colágeno | 0,015 | 0,00 | 0,56 | 0,28 |
| ADP-fosfolípidos | 0,046 | 0,14 | -1,47 | 0,06 |
| Epinefrina-colágeno | 0,015 | 0,00 | 0,56 | 0,28 |
| Colágeno-fosfolípidos | 0,00 | 0,00 | - | - |
| Epinefrina-fosfolípidos | 0,00 | 0,00 | - | - |
| ADP-epinefrina-colágeno | 0,12 | 0,00 | 1,66 | 0,048 |
| ADP-epinefrina-fosfolípidos | 0,046 | 0,047 | 0,01 | 0,49 |
| ADP-colágeno-fosfolípidos | 0,00 | 0,00 | - | - |
| Epinefrina-colágeno-fosfolípidos | 0,00 | 0,00 | - | - |
| ADP-epinefrina-colágeno-fosfolípidos | 0,015 | 0,33 | -4,34 | 7,00e-06 |
| Fosfolípidos como factor común | 0,18 | 0.52 | 2,76 | 0,0056 |
ADP: difosfato de adenosina.
Fuente: Historias clínicas del servicio de genética del Hospital Pediátrico William Soler
Además, la combinación ADP-epinefrina-colágeno-fosfolípidos muestra la diferencia más significativa estadísticamente (p=7,0 y 10-6) asociada al SED. El defecto de liberación en estos casos con fenotipo de Ehlers-Danlos es cualitativamente más complejo que en las personas con HMA, que puede marcar la diferencia en cuanto a la gravedad del endofenotipo.
Al sumar todos los casos con alteraciones de los fosfolípidos de forma aislada o combinada en los casos con HMA (12 pacientes para un 18,46 %) comparados con 11 casos de SED (52,38 %), la diferencia resultó estadísticamente significativa (p=0,0056) (véase la tabla 5).
En general, los pacientes con HMA mostraron asociación mayor con los trastornos en la liberación de gránulos con agonistas como el ADP y epinefrina aislados o combinados y los casos de Ehlers-Danlos con la disponibilidad de los fosfolípidos asociada al resto de los agonistas, diferencias que resultaron estadísticamente significativas con p menor de 0,05.
Discusión
Los trastornos de la fibra colágena son comunes y poco reconocidos como contribuyentes a los síntomas de sangramiento. Los defectos más comunes en las trombopatías, muy frecuentes en la edad pediátrica, son inducidos por epinefrina (32,5 % de positividad), seguidos por los inducidos por ADP (30,5 %) y por los trastornos en la disponibilidad de los fosfolípidos plaquetarios (30 %).
Se estima que hay una alta prevalencia de defectos hereditarios de agregación plaquetaria y que las respuestas al ADP disminuidas con epinefrina y colágeno normal son altamente predictoras de trastornos de función; por eso, en el 60 % de los casos remitidos por sangramiento hay resultados positivos.2
De los casos con hipermovilidad, en un 67 % hubo antecedentes personales o familiares de alteraciones de la fibra colágena, por lo que la hipermovilidad articular sintomática puede asociarse a un trastorno de sangramiento asintomático. En estudios de casos con Ehlers-Danlos hipermóvil se evidenció la existencia de trastornos cualitativos con predominio de la disminución de la agregación con ADP, sola o combinada con epinefrina y colágeno, y con menor frecuencia trastornos de la disponibilidad de los fosfolípidos plaquetarios, al igual que en el presente estudio con algunas diferencias en la composición de la muestra.10,11,22
Vale señalar que clínicamente hay un espectro de casos que van desde la presencia solamente de hipermovilidad y otros que tienen todos los signos clínicos de Ehlers-Danlos, por lo que pudieran haberse incluido algunos casos con variante hipermóvil en el grupo de los Ehlers-Danlos. Debido al periodo en que se realizó el estudio se hace la disquisición entre HMA y SED variante hipermóvil, aunque en la actualidad se consideran una entidad única solo diferenciada por el nivel de afectación de la estructura proteica. LA HMA se consideró una condición diferente del SED variante hipermóvil, aunque no existen hallazgos histopatológicos, ultraestructurales y moleculares patognomónicos que los diferencien. Existe una gran variabilidad de síntomas y signos, pero el asesoramiento clínico en centros especializados es suficiente para confirmar este diagnóstico en el 90 % de los casos.2,10,11,22
En cuanto al mecanismo que puede estar afectado en estos casos estaría la adhesión plaquetaria al colágeno fundamentalmente de los tipos I y III y la formación de enlaces firmes que dependen de la estructura fibrilar del colágeno.17,18,19,20,21,22,23
La fibrilinogénesis es de gran importancia para el desarrollo celular y el colágeno tipo I es un factor ambiental del nicho donde se forman y activan los megacariocitos. Los cambios en sus propiedades mecánicas se manifiestan en respuestas bioquímicas y regulan el comportamiento celular. La función del megacariocito está relacionada con la organización y biomecánica del colágeno, en especial el tipo I, que se relaciona con la rigidez y los patrones espaciales.18,19
En los casos con alteración de la fibra colágena resultaría útil valorar el estudio de función o agregación de plaquetas como elemento endofenotípico que puede contribuir al diagnóstico diferencial y, además, para la prevención de complicaciones que pueden presentarse al extraer piezas dentales o ante cirugías o traumatismos donde se deben administrar medicamentos como el ácido tranexámico y el epsilón amino caproico.
Conclusiones
Los trastornos de agregación plaquetaria pueden presentarse asociados a las alteraciones de la fibra colágena en más de un 90 % de los casos y las alteraciones de los fosfolípidos plaquetarios se presentan con mayor frecuencia en los casos con síndrome Ehlers-Danlos, así como la combinación del trastorno de agregación con todos los factores. Las alteraciones de agregación con ADP y combinación ADP-epinefrina son más frecuentes en la hipermovilidad articular. El estudio de trombopatías puede ser una herramienta para conocer el endofenotipo funcional plaquetario como elemento diferencial en los trastornos de la fibra colágena.