










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
Este es uno de los trastornos más frecuentes inherentes al tejido conectivo. El síndrome de Marfán es una enfermedad autosómica dominante con una incidencia aproximada de 1 a 3000-5000 casos.1 Existe un espectro clínico amplio sobre la gravedad de la enfermedad, desde manifestaciones oculares leves, hasta trastornos cardiovasculares, entre los cuales la disección aórtica aguda es la causa principal de muerte como consecuencia de la degeneración de la capa medial y la formación de aneurismas que comprometen la vida del paciente.1,2
Esta enfermedad se caracteriza por una mutación de la fibrilina 1, una proteína que es codificada por el gen FBN 1, que se encuentra ubicado en el cromosoma 15q21.1. Esta forma parte de uno de los tantos componentes de la matriz extracelular del tejido conectivo y no elásticos. Es la principal proteína de las que conforman las microfibrillas, y que se ocupan de la fibrilogénesis elástica.3
Presentación de caso
Se presenta un paciente masculino de 43 años de edad, de raza negra, admitido en la Clínica Cartagena del Mar en septiembre de 2019 por tos con expectoración verdosa, asociado a dificultad respiratoria desde aproximadamente 20 días de evolución, pero que empeoró en la última semana con la aparición de la tos. En la radiografía de tórax se evidenció un proceso neumónico basal izquierdo por lo cual se decidió hospitalizar.
El paciente es el producto de una segunda gestación por parto natural, eutócico, a término. Fue concebido por su padre a la edad de 33 años y la madre a los 28 años. La estatura del padre es de 1,77 m, actualmente tiene 71 años de edad y es un individuo sano. La madre mide 1,67 m, está viva y es sana. Sus dos hermanos son sanos y ninguno de los dos es de talla alta. Su infancia y desarrollo fueron normales, no se constató deterioro cognitivo y finalizó la educación básica primaria, pero por motivos no asociados a la enfermedad desertó de los estudios. El paciente tiene una hija sana, aunque el padre no estuvo de acuerdo con realizarle estudios para descartar la enfermedad.
Entre los antecedentes de importancia se constató una cirugía de cristalino no especificada por el paciente, que ocurrió 5 años atrás después de un ingreso secundario a pérdida de la agudeza visual en el ojo derecho.
Durante el examen físico al ingreso se halló una tensión arterial de 120/70 mmHg, frecuencia cardiaca de 66 latidos/min, frecuencia respiratoria de 37 respiraciones/min y estaba afebril. El peso fue de 84 kg, talla 1,96 m y envergadura de 2,05 m.
Entre los hallazgos al examen físico encontramos dolicostenomelia (Fig. 1), pectum excavatum, aracnodactilia (Fig. 2), estrías atróficas, signo de Walter Murdoch positivo y signo de Gowers (Fig. 3), paladar ojival (Fig. 4), escoliosis con cavidad derecha de vértebras torácicas (Fig. 5), alta talla y soplo diastólico en foco aórtico III/VI.
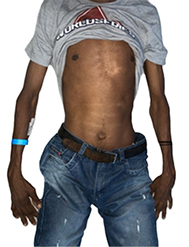
Fig. 1 Se observa dolicostenomelia que es el alargamiento de los miembros en relación con el tronco.
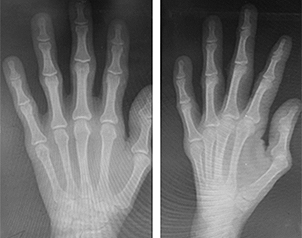

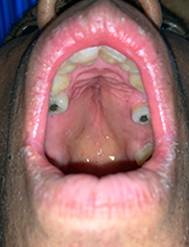
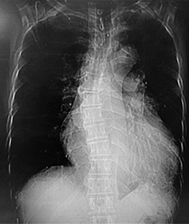
Fig. 5 Escoliosis de concavidad derecha de las vértebras torácicas; se observa, además, gran cardiomegalia.
Los exámenes de laboratorio del paciente evidenciaron una hemoglobina de 13,2 g/dL, hematocrito 42,4 %, volumen corpuscular medio 100,6 fl, hemoglobina corpuscular media 31,2 pg, leucocitos, 9500 células/mm3, neutrófilos 80 %, linfocitos 15 %, eosinófilos 5 %, conteo de plaquetas 171 000 U/mm3, y proteína C reactiva 24 mg/L.
En el ecocardiograma transtorácico, se observó la dilatación aneurismática del anillo valvular aórtico y seno de Valsalva, insuficiencia valvular aórtica grave y prolapso de la válvula mitral.
En la angiotomografía de tórax se observó la dilatación de la raíz de la aorta ascendente, con insuficiencia valvular aórtica secundaria.
Durante la estancia del paciente se trató su cuadro de neumonía adquirida en la comunidad con piperacilina/tazobactam por 7 días. Se indicó la valoración de un especialista en cirugía cardiovascular por el hallazgo de un aneurisma de la aorta ascendente, pero el paciente decidió no continuar con los estudios de su enfermedad. Se dio de alta al paciente concluido el tratamiento para la neumonía y se hicieron recomendaciones para lograr cambios en el estilo de vida, la práctica de ejercicios físicos y se insistió en la alta probabilidad de muerte debido al problema vascular descrito.
Discusión
El síndrome de Marfán es una enfermedad hereditaria que generalmente no se manifiesta en la edad temprana; luego durante la adolescencia y después de los 20 años se empiezan a observar las alteraciones esqueléticas, algunas oculares como la disminución temprana de la agudeza visual y los síntomas cardiovasculares dados por el prolapso de la válvula mitral o los trastornos de la válvula y raíz aortica.1
A pesar de ser una enfermedad autosómica dominante, existe un porcentaje (5-35 %) de los casos en los cuales la enfermedad aparece de forma esporádica y en los que se pueden presentar nuevas mutaciones cromosómicas que generen una expresividad diferente de la enfermedad.4
Este síndrome se diagnostica fundamentalmente a partir de la presencia de signos clínicos que fueron descritos sobre la base de los criterios de Ghent expuestos en 1986, modificados más adelante en 2010, catalogados como los criterios de Ghent modificados en los que se da más peso a las alteraciones de la raíz aórtica: disección o aneurisma y ectopia lentis (Tabla).5
En ausencia de antecedentes familiares de la enfermedad, se puede establecer el diagnóstico de la enfermedad de Marfán de varias formas:
Criterio aórtico (diámetro de la línea Z mayor que 2 o igual, o disección aórtica) y ectopia lentis.
Criterio aórtico (diámetro de la línea Z mayor que 2 o igual, o disección aórtica) y una mutación del FBN 1.
Criterio aórtico (diámetro de la línea Z mayor que 2 o igual, o disección aórtica) y más de 7 puntos en la puntuación de manifestaciones sistémicas de la escala de Ghent modificada.
Presencia de ectopia lentis y mutación del FBN 1 que se ha identificado con la lesión aórtica aneurismática.
Tabla Criterios sistémicos de la clasificación de Ghent modificada. La presencia de 7 o más criterios sistémicos confirman el compromiso sistémico de la enfermedad, acompañado de la presencia o no de antecedentes familiares
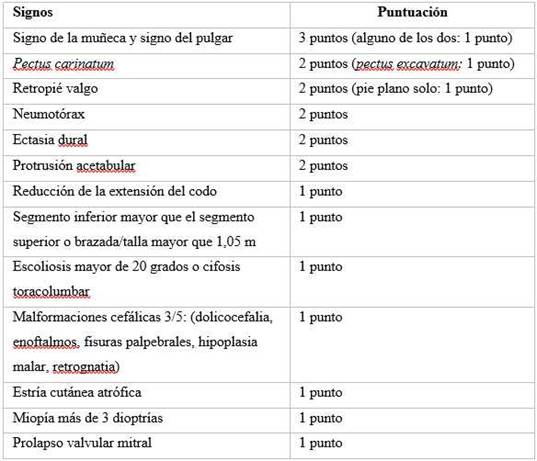
Fuente: (5) Loeys BL, et al. The revised Ghent nosology for the Marfan syndrome. J Med Genet. 2010;47(7):476-85.
Si hay antecedentes de la enfermedad, mediante la presencia de alguno de estos criterios se establece el diagnóstico de la enfermedad:
Ectopia lentis.
7 puntos o más de manifestaciones sistémicas en la escala de Ghent modificada.
Criterio aórtico (diámetro de la línea Z mayor que 2 o igual en mayores de 20 años, o un puntaje de línea Z mayor que 3 o igual en menores de 20 años, o la presencia de disección aórtica).
Se debe tener en cuenta que en este último criterio tiene que estar ausente la sospecha diagnóstica de enfermedades como el síndrome de Loeys-Dietz, Ehlers-Danlos y el síndrome de Shprintzen-Goldberg.5
El paciente estudiado presentaba varios de los criterios para el diagnóstico de la enfermedad. Si hacemos el ejercicio de aplicar los criterios de Ghent modificados encontramos que el paciente presentaba dilatación aneurismática de la aorta, signo de la muñeca y del pulgar, pectum excavatum, escoliosis, malformaciones cefálicas que consistían en dolicocefalia, retrognatia, estrías cutáneas atróficas, disminución de la extensión del codo, dolicostenomelia y el antecedente de una cirugía oftalmológica por ectopia lentis desde hacía 5 años, por lo que determinamos una alta sospecha diagnóstica de la enfermedad.
Los diagnósticos diferenciales más frecuentes en este tipo de pacientes son la homocisteinuria (que suele causar retraso mental y trastornos tromboembólicos); el fenotipo MASS que viene del acrónimo en inglés Mitral prolapse, Aortic disease, Stria atrophic, Skeletical, pero en este fenotipo no aparece la ectopia lentis, antecedente que sí tenía nuestro paciente; en el síndrome de Loeys-Dietz que es otro de los diagnósticos diferenciales es un criterio observar la úvula bífida6,7 que nuestro paciente no presentaba; y el síndrome de Ehlers-Danlos no cursa con altura desproporcionada, pectum carinatum o dilatación progresiva de la aorta. Por lo tanto, clínicamente podíamos llegar a pensar en el síndrome de Marfán.
Para finalizar, el tratamiento del síndrome de Marfán consiste en la corrección de las manifestaciones sistémicas que presente el paciente; desde el punto de vista de las malformaciones esqueléticas se puede realizar la corrección del pectum carinatum o excavatum, la corrección del retropié valgo, ortodoncia para el apilamiento dental, la faquectomía en caso de la ectopia lentis y el uso de gafas médicas si el paciente presenta miopía. Las otras alteraciones en el sistema cardiovascular que tienen un pronóstico más grave deberían seguirse periódicamente por un especialista de cardiología y cirugía cardiovascular para evitar la muerte, prever las complicaciones y realizar tratamiento quirúrgico electivo urgente o emergente dependiendo el caso.
El tratamiento farmacológico con betabloqueantes ha demostrado una disminución en la progresión de la dilatación del seno de Valsalva. El propanolol fue el primer medicamento al cual se le atribuyó este beneficio; otros betabloqueantes como el metropolol o el carvedilol han demostrado poder disminuir la complicación.8,9 También se ha evidenciado la reducción del anillo aórtico en pacientes que usan antagonistas de los receptores de la angiotensina II (ARA II) como losartán, ya que algunos pacientes no toleran los efectos adversos de los betabloqueantes, y los anteriores también disminuyen la progresión de la dilatación aneurismática.10 Cabe resaltar, además, que los bloqueadores de los canales de calcio están contraindicados en este tipo de pacientes, ya que aumentan el riesgo de disección y la necesidad de cirugía aortica.11
Respecto al tratamiento quirúrgico de la vasculopatía aórtica hay estudios que soportan por obvias razones la corrección electiva de la enfermedad aórtica de forma electiva, pues se asocian con una tasa menor de mortalidad a los 30 días, cuando se realiza la cirugía de forma electiva, urgente o emergente. Estos fueron los tres grupos que se compararon, apoyando la realización de la cirugía electiva: indicaciones para el reparo aórtico que incluye un diámetro mayor de 50 mm; crecimiento de la dilatación acelerado (más de 5 mm por año); antecedente familiar de disección aórtica en pacientes con diámetros menores de 50 mm o presencia de insuficiencia valvular aórtica progresiva.12
En cuanto a la técnica quirúrgica para el reparo aórtico, no se encontraron diferencias entre la técnica de Davies y la técnica de Bentall respecto a la mortalidad a los 30 días.12

