










 Servicios personalizados
Servicios personalizados
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
Introducción
La poliangeítis microscópica es un subtipo de vasculitis asociada a anticuerpos anticitoplasma de neutrófilos (ANCA). Se define como una enfermedad autoinmune, multisistémica, caracterizada por presentar vasculitis necrotizante, con ausencia o escasa presencia de inmunocomplejos, y que afecta predominantemente a vasos de pequeño calibre (capilares, vénulas o arteriolas) con ausencia de inflamación granulomatosa.1
Su incidencia global anual es de 0,2-1,5 casos por cada 100 000 habitantes, con un pico en la séptima década de vida, sin predominio de ningún sexo. Existe una gran variación de acuerdo con cada región geográfica y las diferencias étnicas, y hay una mayor incidencia en Asia, Oceanía y en el sur de Europa.2,3,4,5
Su grado de afectación clínica es variable e impredecible: desde alteraciones mínimas hasta manifestaciones que ponen en riesgo la vida del paciente. El órgano más frecuentemente afectado es el riñón, en un 95,3 % de los casos, seguido de la enfermedad pulmonar (56,6 %), afectación cutánea (22,6 %), neurológica (18,9 %) y articular (7,1 %).2,3 La hemorragia alveolar difusa, la tasa de filtrado glomerular <15 mL/min/1,73 m2, la afectación neuronal, la edad mayor de 65 años y la presencia de anticuerpos antimieloperoxidasa (anti-MPO) se encuentran relacionados con una mayor mortalidad, por lo que se consideran predictores de mal pronóstico.5,6,7
Con la sospecha clínica de vasculitis de pequeños vasos, para llegar al diagnóstico definitivo es necesario realizar exámenes complementarios en donde se constate la presencia de ANCA de patrón perinuclear o citoplasmático. En la poliangeítis microscópica el patrón anti-MPO está presente en un 82,1 %; un 8,5 % de los casos son positivos para anticuerpos anti-proteinasa 3 (anti-PR3) y un 5,7 % son doble positivo, por lo que debe asociarse al resultado histopatológico del tejido afectado, en este caso con presencia de glomerulonefritis pauciinmune crescéntica.2,3,8
El tratamiento recomendado actualmente comprende una dosis de carga intravenosa de glucocorticoides y terapia de inducción para la remisión con potentes drogas inmunosupresoras, las que a menudo presentan riesgo de efectos adversos significativos (ciclofosfamida, rituximab o metotrexato), junto a profilaxis contra Pneumocystis jirovecii (antiguamente conocido como P. carinii). Adicionalmente, es posible utilizar terapia de intercambio de plasma sanguíneo, si hay glomerulonefritis rápidamente progresiva, especialmente en caso de creatinina sérica mayor de 5,7 mg/dL o hemorragia alveolar difusa.5,9
El porcentaje de remisión en pacientes tratados con ciclofosfamida y glucocorticoides es del 85-90 % en 2 a 6 meses;10 con una tasa estimada de supervivencia de 98,6 %, 89,6 %, 75,1 % y 60,4 % luego de 1; 5; 10 y 20 años, respectivamente. Un 25,5 % de los pacientes poseen riesgo de recaída, en un tiempo medio entre 17-38,9 meses.3,7,11
Para el diagnóstico diferencial de esta enfermedad se debe considerar principalmente las afecciones que se manifiestan como síndrome pulmonar-renal, las que se clasifican en cuatro grupos en orden descendente de incidencia:
Vasculitis sistémicas (granulomatosis con poliangeítis, poliangeítis microscópica y granulomatosis eosinofílica).
Enfermedad anti-membrana basal glomerular.
Enfermedades autoinmunes (lupus eritematoso sistémico, crioglobulinemia).
Microangiopatías trombóticas (infecciones, neoplasias).12
Caso clínico
Se presenta un paciente masculino de 49 años, mestizo, agricultor, sin antecedentes patológicos de importancia, quien refirió un cuadro clínico de 30 días de evolución, con presencia de tos seca de tipo irritativa, que evolucionó a tos productiva, hemoptisis, disnea de moderado esfuerzo, astenia, artralgias y mialgias. Asociado al cuadro clínico refirió edema de miembros inferiores de 7 días de evolución. Al examen físico, se observa palidez generalizada, taquipnea, taquicardia, temperatura corporal de 38,3 °C, aliento urémico, campos pulmonares hipoventilados, estertores crepitantes escasos en las bases pulmonares, extremidades inferiores con presencia de edema ++ que deja fóvea +.
Mediante los exámenes complementarios se constata anemia grave, normocrómica, normocítica, hiperpotasemia, tasa de filtrado glomerular de 6,9 mL/min/1,73 m2, uroanálisis con presencia de sangre +++, proteínas +, proteínas en orina de 24 h de 238,42 mg. Posteriormente, con la sospecha diagnóstica establecida de vasculitis de pequeños vasos, se realizan exámenes confirmatorios, en donde se obtiene presencia de ANCA positivo, de tipo p-ANCA, sistema de complemento dentro de los parámetros normales y negatividad de anticuerpos antimembrana basal glomerular (anti-MBG) (Tabla).
Tabla- Resultados de los exámenes complementarios realizados al paciente con sospecha de vasculitis de pequeños vasos
| Exámenes generales | |||||
| Glóbulos blancos: | 6,100 | Glóbulos rojos | 2,200 | Urea | 209,7 mg/dL |
| Neutrófilos: | 73 % | Hemoglobina | 6,1 g/L | Creatinina | 8,18 mg/dL |
| Linfocitos: | 18 % | Hematocrito | 19,4 % | PCR | 0,3 mg/L |
| Monocitos: | 5,9 % | VCM | 87,8 fL | Sodio | 138 mmol/L |
| Eosinófilos: | 0,19 % | HCM | 29 pg | Potasio | 6,55 mmol/L |
| Plaquetas: | 266 000 | CHMC | 33 g/dL | Cloro | 105 mmol/L |
| Estudios inmunológicos | |||||
| ANA | Negativo | ANCA | Anti-MPO: 253 U/mL | Anti-MBG | Negativo |
| Complemento C3 | 100 mg/dL | Complemento C4 | 39 mg/dL | Coombs | Negativo |
Leyenda: VCM: volumen corpuscular medio; HCM: hemoglobina corpuscular media; CHMC: concentración de hemoglobina corpuscular media; PCR: proteína C reactiva; ANA: anticuerpo antinuclear; ANCA: anticuerpo anticitoplasma de neutrófilos; Anti-MPO: anticuerpos mieloperoxidasa; Anti-MBG: anticuerpo antimembrana basal glomerular.
Fuente: Historia Clínica Unificada.
Conjuntamente se solicita estudios imagenológicos de tórax y abdomen. Se observan infiltrados de tipo difuso con aspecto de vidrio deslustrado altamente sugestivos de hemorragia difusa a nivel alveolar. A nivel abdominal se identifican los riñones de aspecto y tamaño normal, con relación corticomedular conservada (Fig. 1 y Fig. 2).
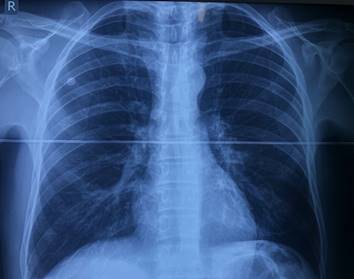
Fig. 1- Radiografía posteroanterior de tórax, donde se observan infiltrados pulmonares de tipo difuso con aspecto de vidrio deslustrado.

Fig. 2- Tomografía de tórax. Se aprecian múltiples áreas coalescentes de patrón en vidrio deslustrado y áreas de patrón de perfusión en mosaico con predominio en las bases pulmonares.
Evolución
Considerando la presencia de glomerulonefritis rápidamente progresiva, enfermedad pulmonar intersticial, hemorragia alveolar difusa y ANCA de tipo anti-MPO positivo, se diagnostica un cuadro clínico altamente sugestivo de poliangeítis microscópica, con un puntaje de 21 de acuerdo con la tercera versión de la Escala de Birmingham para la actividad de vasculitis (BVAS, por sus siglas en inglés). Se decide iniciar tratamiento con dosis de carga de 1 g diario de metilprednisolona por 3 días, seguido de pulsos de ciclofosfamida con una dosis de 0,6 g/m2 (1 g) hasta cumplir 6 dosis y hemodiálisis trisemanal; se incluye profilaxis antibiótica contra P. jirovecii con trimetopim-sulfametoxazol a 800/160 mg y 2-mercaptoetanol sulfonato de sodio (MESNA) para la prevención de cistitis hemorrágica. El paciente presentó posteriormente la remisión del cuadro; sin embargo, debido al daño tisular permanente ocasionado a nivel renal, no existió mejoría en la tasa de filtrado glomerular, por lo que se mantiene en terapia de sustitución renal permanente, con una frecuencia trisemanal.
Discusión
Una vez establecida la sospecha de vasculitis de pequeños vasos: poliangeítis microscópica, la piedra angular para establecer el diagnóstico definitivo, se basa en la presencia de ANCA (perinuclear-citoplasmático), negatividad de anticuerpos anti-MBG, junto con la biopsia del tejido que demuestre vasculitis a nivel histológico, siempre que esta sea posible o con estudios de imagen que muestren lesiones sugestivas en casos en los que no se puede realizar el estudio histopatológico.6,13 Por esa razón en el presente caso, debido a las manifestaciones que ponían en riesgo la vida del paciente, se dejó en segundo plano los estudios histopatológicos, ya que la presencia de ANCA positivo en pacientes con glomerulonefritis rápidamente progresiva posee un valor predictivo positivo del 98 % para glomerulonefritis pauciinmune crescéntica en biopsias renales.14
Cabe recalcar lo controversial del inicio del tratamiento en pacientes que no poseen biopsia renal confirmatoria debido a su toxicidad, por lo que esta se debe solicitar de manera personalizada, según el cuadro clínico y la estabilidad del paciente. Varias fuentes recomiendan realizar biopsia de tejido únicamente en los casos en los que el contexto clínico, la valoración física y los exámenes complementarios no identifiquen la causa del daño renal, debido al alto riesgo de hemorragia, disminución de la viabilidad renal y morbilidad y mortalidad asociadas al procedimiento.15
El diagnóstico y tratamiento de forma precoz disminuyen considerablemente la morbilidad y mortalidad, especialmente la necesidad de terapia de reemplazo renal permanente, que se presenta en un 34 % de los casos. En el caso de nuestro paciente, a pesar de que el tiempo desde el inicio del cuadro clínico hasta el inicio del tratamiento fue de aproximadamente 30 días y con remisión posterior al tratamiento, el haber presentado hemorragia alveolar difusa, junto a la necesidad de terapia de reemplazo renal y la presencia de anticuerpos anti-MPO inclinan hacia un mal pronóstico, con un mayor riesgo de recaídas y mortalidad, especialmente durante el primer año.
Conclusiones
La poliangeítis microscópica, a pesar de ser una enfermedad de rara incidencia, posee una llamativa morbilidad y mortalidad, por lo que es trascendental el conocimiento de su presentación clínica, procedimiento diagnóstico y tratamiento, con el fin de obtener un impacto positivo en la calidad de vida y la supervivencia del paciente. Su pronóstico depende principalmente del diagnóstico precoz y evitar posibles daños irreversibles en los órganos afectados.
Existe una gran variación en la incidencia de las vasculitis asociadas a ANCA, de acuerdo con la distribución geográfica y las diferencias étnicas, lo que, sumado a una falta de estudios clínicos y epidemiológicos en nuestra población latinoamericana, tornan el diagnóstico de esta enfermedad muy difícil. La descripción de casos de este tipo de enfermedades estimula la investigación local y regional, de gran importancia dentro de la salud pública.

