










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
Una gammapatía monoclonal siempre es el resultado de una proliferación de células clonales.1 Existen entidades ampliamente reconocidas en asociación a las discrasias de células sanguíneas. Entre los sistemas que se afectan con mayor frecuencia están el sistema nervioso, tanto central como periférico como ocurre en la polineuropatía del síndrome de POEMS; el renal (gammapatías de significado renal) con necrosis tubular por el depósito de paraproteínas, por ejemplo en la amiloidosis y la piel con mecanismos más complejos y variados.
Lipsker y otros1 proponen el término “gammapatías de significado cutáneo” y organizan las afectaciones según el mecanismo patogénico en: 1) depósito de inmunoglobulinas (Ig) o paraproteínas (vasculares o extravasculares); 2) actividad biológica de inmunoglobulinas (xantogranuloma necrobiótico); 3) secreción anormal de citocinas (síndromes de POEMS y Schnitzler); 4) infiltración de células plasmáticas (plasmocitomas a distancia); y 5) desconocido, como la cutis laxa.
Este amplio espectro de manifestaciones puede darse antes del diagnóstico de la gammapatía, por lo cual es de suma importancia su conocimiento ya que pueden simular enfermedades reumáticas. El propósito de este estudio es comunicar los casos de cinco pacientes con manifestaciones clínicas de enfermedades reumáticas y diagnóstico final de enfermedades oncohematológicas.
Métodos
Estudio descriptivo transversal a través del análisis de historias clínicas de pacientes evaluados en el Servicio de Reumatología del Hospital José María Cullen de Santa Fe entre marzo del 2010 y junio del 2019. Se incluyó a cinco pacientes que fueron estudiados por sospecha de enfermedad reumatológica hasta llegar al diagnóstico final de gammapatía monoclonal.
Casos clínicos
Cuatro de los pacientes estudiados presentaron mieloma múltiple y uno linfoma intravascular (LIV). Entre los cuadros clínicos encontramos un paciente con síndrome de Schnitzler; uno con xantogranuloma asociado a amiloidosis; aplastamientos vertebrales múltiples; falla renal aguda y el último con una afectación multisistémica con sospecha de vasculitis primaria.
Caso 1: Paciente femenina de 38 años, sin antecedentes de salud que consulta por urticaria crónica caracterizada por lesiones maculopapulares en el tronco y la raíz de los miembros superiores, no dolorosas ni pruriginosas (Fig. 1) y perforación del tabique nasal. Durante su evolución presentó dolor abdominal, diarrea, hepatoesplenomegalia, dolor óseo, parestesias, livedo reticularis e hipoacusia neurosensorial rápidamente progresiva. En los exámenes de laboratorio se halló una banda monoclonal IgG y en la biopsia de piel se informó vasculitis leucocitoclástica. Se diagnosticó síndrome de Snichtzler. La paciente desarrolló un mieloma múltiple 9 años después.
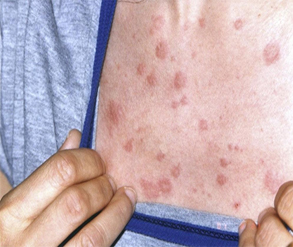
Fig. 1 Lesiones maculopapulares en el tórax propias del síndrome de Snichtzler, correspondientes al caso clínico 1.
Caso 2: Paciente masculino de 51 años con antecedente de mieloma múltiple desde hacía 13 años tratado en controles periódicos por la especialidad de hematología. Consultó en esta ocasión por edema de párpados bilateral, de coloración amarillenta, de 3 años de evolución que agregó equimosis palpebral bilateral y ulceración del lado derecho. Se realizó una primera cirugía para extirpar xantelasmas sin estudio de la pieza quirúrgica. Posteriormente reaparecieron las lesiones en mayor grado, momento en el cual se evidenció por anatomía patológica macrófagos cargados de lípidos, sin necrobiosis y tinción positiva con rojo Congo. Se diagnosticó xantogranuloma del adulto y amiloidosis (Fig. 2).
Caso 3: Paciente femenina de 35 años con dolor lumbar de 3 meses, tipo inflamatorio, sin síntomas neurológicos. En las imágenes radiográficas (Fig. 3) presentó aplastamientos vertebrales múltiples, laboratorio con pico gamma monoclonal de tipo IgG, parámetros de metabolismo fosfocálcico normal. Se realizó biopsia de médula ósea compatible con mieloma múltiple. Después del diagnóstico esta paciente fue derivada al Servicio de Oncología para su atención.
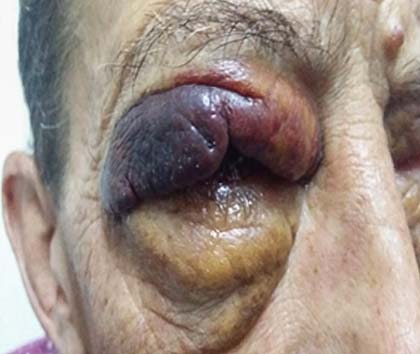
Fig. 2 Lesión tumefacta purpúrica y ulcerada en el párpado derecho en el paciente correspondiente al caso clínico 2.

Caso 4: Paciente masculino de 61 años con antecedentes de vasculitis leucocitoclástica en la piel y adenocarcinoma prostático, que comenzó con síntomas respiratorios y digestivos inespecíficos, hipertensión arterial, edemas periféricos y derrame pleural, compromiso renal (proteinuria de 3 g) y hemorragia cerebral, por lo que es derivado por sospecha de vasculitis. En los estudios complementarios se halló plaquetopenia, anemia leve, función renal normal, ferritina mayor de 1000 ng/mL; el proteinograma por electroforesis en sangre determinó hipoalbuminemia, hipogammaglobulinemia grave, aumento de B2 microglobulina, presencia de componente monoclonal IgM de la cadena liviana kappa y electromiograma con polineuropatía mixta. En la punción de médula ósea se observó la población celular con inmunomarcación compatible con un proceso linfoproliferativo B clonal. Aparecieron, además, lesiones purpúricas en piel (Fig. 4), por lo cual se le realizó biopsia de las lesiones y del nervio sural donde se evidenció LIV, por lo cual fue derivado al Servicio de Hematología.
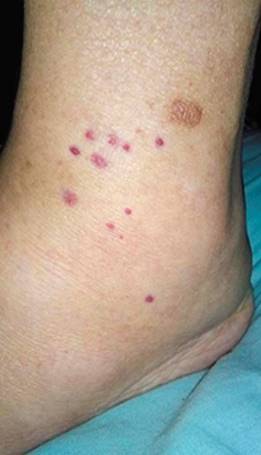
Caso 5: Paciente femenina de 46 años que ingresó por falla renal rápidamente progresiva, con oligoanuria y requerimiento de diálisis. Dos meses antes de la consulta refirió evidenciar disminución del ritmo diurético. En los estudios de laboratorio se detectó FAN positivo a títulos 1/640 con patrón homogéneo, por lo cual se nos solicitó la valoración por sospecha de glomerulonefritis lúpica. Se realizó biopsia renal y posteriormente pulso de esteroides. En el informe de anatomía patológica se describió depósito de cadenas livianas por lo cual se iniciaron los estudios de gammapatía. Se evidenció un 7 % de células plasmáticas en microscopía óptica con citometría compatible con mieloma múltiple y se repitió luego con recuento superior al 10 %, por lo cual se derivó a hematología para iniciar tratamiento específico.
Discusión
Las discrasias de células sanguíneas engloban trastornos proliferativos de diferentes estirpes. Entre las neoplasias de linfocitos B se encuentran, además de linfomas, las gammapatías como el mieloma múltiple, la gammapatía monoclonal de significado incierto y la amiloidosis por cadenas livianas.2
La proliferación clonal de células plasmáticas da como resultado la secreción del llamado componente “M”, monoclonal o paraproteína, que puede ser formada íntegramente de Ig o de alguna porción (cadenas pesadas o livianas), o una combinación de ambas. Presentan la característica imagen de “pico monoclonal” en el proteinograma por electroforesis, con bandas monoclonales en sangre y orina, que siempre deben solicitarse ante la sospecha de estas enfermedades.2 Es interesante el desarrollo de los mecanismos fisiopatológicos de las diferentes manifestaciones generadas por estas entidades que como mencionamos pueden ser por depósito de Ig o paraproteínas de forma vascular (crioglobulinemia I) o extravascular (amiloidosis); por actividad biológica de Ig (xantogranuloma necrobiótico); por secreción anormal de citocinas (síndrome de POEMS o Schnitzler); por infiltración de células plasmáticas (plasmocitomas a distancia); o por mecanismos desconocidos como la cutis laxa.1
Esta serie fue muy interesante, ya que estos pacientes llegaron a la consulta por sospecha de enfermedades reumatológicas, lo cual debemos tener presente para evitar tratamientos inmunosupresores innecesarios.
La paciente 1 con el síndrome de Schnitzler desarrolló un mieloma múltiple 9 años después. Esta entidad se considera un síndrome autoinflamatorio adquirido, dentro de las inflamosomopatías. Se presentó mucho antes del diagnóstico de mieloma múltiple con urticaria crónica y síntomas sistémicos que simulaban una vasculitis, pero después con el proteinograma por electroforesis se evidenció la banda monoclonal por IgG kappa (aunque es característica la IgM).3,4
Desde el punto de vista fisiopatológico ocurre por un aumento de la IL-1B, aunque se desconoce si el componente monoclonal es una causa o una consecuencia de esta liberación.
Este cuadro clínico se encuentra como diagnóstico diferencial de la urticaria crónica junto con otras entidades como la crioglobulinemia, vasculitis urticariana hipocomplementémica, deficiencia adquirida de inhibidor C1, síndrome hiper-IgD y enfermedad de Still del adulto.5
Existen criterios diagnósticos desarrollados por Dan Lipsker5 (Tabla), que consisten en dos criterios mayores (ambos requeridos): exantema urticariano crónico y gammapatía monoclonal IgG o IgM;6 y al menos dos criterios menores: fiebre intermitente, artralgias/artritis, dolor óseo, linfoadenopatía, hepatoesplenomegalia, aumento de la velocidad de sedimentación globular (VSG), leucocitosis y anormalidades óseas (confirmadas por imagen o histología).
El tratamiento de este síndrome era infructuoso hasta el advenimiento de los inhibidores de la IL-1B como el canakinumab.7 El caso de esta paciente fue publicada previamente por nuestro grupo.8
Tabla- Criterios diagnósticos de Dan Lipsker para el síndrome de Schnitzler
| Criterios mayores | Críticos menores |
|---|---|
|
|
Fiebre intermitente Artritis o artralgia Dolo óseo Linfoadenopatía palpable Esplenomegalia o hepatomegalia VSG acelerada Leucocitosis Anormalidades óseas (radiográficas o histológicas) |
Nota: Se requieren los dos criterios mayores y al menos dos menores para el diagnóstico de síndrome de Schnitzler.
El paciente 2, también con manifestaciones en la piel, presentó dos trastornos asociados a mieloma múltiple, pero no relacionados entre sí. Inicialmente manifestó una lesión de tipo xantogranulomatosa y luego una amiloidosis. Con respecto a la primera parece oportuno llamar la atención sobre las denominadas “lesiones xantomatosas”,9 granulomas con macrófagos cargados de lípidos que pueden corresponder a dislipidemias de diferentes tipos, causas idiopáticas, o asociadas a enfermedades específicas como la histiocitosis; y en nuestro caso a las gammapatías monoclonales. Es sorprendente la poca importancia atribuida a las lesiones xantomatosas en los párpados en la práctica médica, evidenciada por la falta de estudio histopatológico de la pieza quirúrgica. En la segunda muestra evidenciamos macrófagos cargados con lípidos, pero sin necrobiosis. Dentro de este síndrome de afectación ocular y periocular, las gammapatías toman su lugar, generando mediante la Ig monoclonal actividad de anticuerpos contra la rivoflavina que darán xantelasmas y xantogranulomas, de estos uno de los más frecuentemente hallados es el xantogranuloma necrobiótico caracterizado clínicamente por ulcerarse y anatomopatológicamente por necrobiosis.10,11 La amiloidosis asociada a gammapatías puede generar pápulas, placas, nódulos, engrosamiento de la piel (escloredermia-like), y muchas veces síntomas sugestivos como el fácil sangrado ante los aumentos de presión (maniobra de Valsalva) o por contacto mínimo.12 Las lesiones purpúricas son particularmente frecuentes en la cabeza y el cuello, y pueden evidenciarse en más del 16 % de los pacientes con amiloidosis.11 En nuestro paciente el antecedente de mieloma múltiple sumado al tipo de lesión y coloración con rojo Congo nos dio la certeza diagnóstica (véase la fig. 2).
El caso 3 ilustra a una paciente joven con lo que parecía ser una lumbalgia inflamatoria, por lo cual se iniciaron los estudios para su diagnóstico. Estos presentaban aplastamientos vertebrales múltiples en los estudios radiográficos (véase la fig. 3). Las dos causas más frecuentes de aplastamiento vertebral son la osteoporóticas y la neoplásica.13 En el mieloma múltiple, el dolor lumbar aparece en el 60 % de los pacientes en el momento del diagnóstico, brusco (por microfracturas) o crónico y precipitado por los movimientos. Las lesiones líticas pueden encontrarse casi en el 70 % de los pacientes y el 20 % presenta osteoporosis, fracturas patológicas y vertebrales.14 Si bien hay ciertos indicios imagenológicos de fracturas metastásicas, existen comunicaciones que sugieren que la apariencia benigna en la resonancia magnética nuclear es una de las formas de aplastamiento vertebral en el mieloma múltiple, indistinguible de la causa osteoporótica.15,16
Fisiopatológicamente, la enfermedad ósea en el mieloma múltiple se da por la expansión de células plasmáticas en la médula ósea con destrucción del hueso. Estos pacientes tienen un remodelado óseo anormal por incremento de los osteoclastos estimulados, adyacentes a los cuales se ubican las células mielomatosas, y una disminución de la formación por los osteoblastos, lo que genera lesiones puramente líticas.17 Cabe resaltar que dentro de la afectación ósea del mieloma encontramos los huesos que presentan actividad de la médula ósea en el adulto como el cráneo, las vértebras, las costillas y la porción proximal de los huesos largos, por lo cual son estos los lugares propensos a las fracturas patológicas.
En el caso de nuestra paciente el dolor inició después de la anestesia raquimedular por cesárea, con lo cual entraría dentro del grupo de pacientes en que se desencadena luego de un trauma leve o dolor espontáneo,18 para diferenciarlo de la lesión osteoporótica en que suele ser con traumas de mayor intensidad o de la lumbalgia inflamatoria de comienzo insidioso.
La radiografía simple continúa siendo el método de estudio inicial en la sospecha de lesiones óseas.14
En el caso 4 observamos una enfermedad diferente a las anteriores. Se trata de una proliferación de células B clonales, un linfoma atípico denominado LIV. Fue definido por la Organización Mundial de la Salud como linfoma de células B extranodal, difuso, con afectación solo intraluminal de pequeños vasos, principalmente capilares.19 El diagnóstico es difícil y muchas veces post mortem. Se han descripto dos variantes fenotípicas según la afectación: sistema nervioso central y piel (o solo piel); y multiorgánica con falla sistémica y síndrome hemofagocítico, pero su presentación es heterogénea. Su incidencia es menor de 1 caso por millón de habitantes, y se da entre los 60 y 70 años.20
Los signos más frecuentemente hallados son fiebre (45 %), muchas veces como fiebre de origen desconocido, asociados a deterioro del estado general y pérdida ponderal. La afectación cutánea en un tercio de los pacientes consiste en lesiones que van desde placas induradas hasta descamación y lesiones purpúricas. El compromiso del sistema nervioso es frecuente (40 %) en forma de accidente cerebrovascular, sangrados y convulsiones. Aun así, cualquier órgano puede estar comprometido.
En nuestro caso como datos orientadores hallamos ciertos marcadores de laboratorio a tener en cuenta: anemia, aumento de LDH, aumento de B2 microglobulina y más específicamente el aumento de la ferritina y la hipogammaglobulinemia grave. Este último dato es importante debido a que en la mayor parte de los casos se debe a trastornos hematológicos y no reumáticos. En el caso de las gammapatías se debe a la menor producción de anticuerpos normales y a la mayor destrucción por células reguladoras y aumento de la tasa catabólica de la Ig dependiente de la concentración.2 El diagnóstico definitivo se obtiene mediante anatomía patológica al evidenciar linfocitos atípicos limitados a luz de los pequeños vasos.
Por encontrarse en estadios avanzados en el momento del diagnóstico, en la mayoría de los pacientes el pronóstico es ominoso.
La paciente 5 presentó una falla renal rápidamente progresiva y FAN positivo, lo cual dio lugar a confusión. La falla renal asociada a mieloma múltiple se evidencia hasta en el 20 % de los pacientes al diagnóstico de la enfermedad y hasta el 50 % puede desarrollarlo en su evolución.2 Fisiopatológicamente, el daño que se genera por la toxicidad de las cadenas livianas monoclonales al depositarse en forma de cilindros (riñón de mieloma) es la causa más frecuente en mieloma múltiple (90 %).21 Puede encontrarse en la biopsia inflamación y fibrosis tubulointersticial, asociada a cilindros de cadenas livianas y proteína de Tam Horsfall como el caso de nuestra paciente. El resto de las alteraciones renales se da por mecanismos menos concretos como la reabsorción masiva de cadenas livianas con estrés oxidativo intracelular, medicamentoso o metabólico (por ejemplo, hipercalcemia). Hasta en un 50 % la falla renal es de carácter agudo y habitualmente asociado a un desencadenante.
El diagnóstico se realizó mediante punción/aspiración de la médula ósea con conteo de células por encima del 10 % en la segunda muestra, tal vez debido al curso de esteroides que recibió.
En cuanto al tratamiento de esta complicación uno de los puntos es la remoción directa de cadenas livianas mediante la diálisis,21,22 que probablemente sumado al curso de esteroides que recibió la paciente, haya llevado a su mejoría antes del diagnóstico real.
Nuestra paciente se encuentra actualmente en tratamiento por el Servicio de Hematología, con diuresis espontánea y sin requerimiento de diálisis.
Conclusiones
La similitud de las manifestaciones de algunas enfermedades oncohematológicas y las enfermedades reumáticas obliga a tener en cuenta las características clínicas y analíticas que harán sospechar este grupo de enfermedades y llegar a su rápido diagnóstico.
Como propone Lipsker parece ser que el grupo de gammapatías monoclonales de significación clínica es mucho más amplio de lo que se piensa y difícil de reconocer en los pacientes en los cuales estas manifestaciones aparecen antes del diagnóstico final.

