










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
La esclerodermia es conceptualizada como una enfermedad con componente inmunitario, caracterizada por un aumento de la producción de colágenos que condiciona fibrosis en la piel, tejidos blandos y en distintos órganos. La afectación puede localizarse superficialmente o afectar tejido celular subcutáneo y las capas más profundas de la piel. De forma general se describen varias formas de presentación: esclerodermia localizada (EL), sistémica, síndromes esclerodermiformes y síndromes de overlap o superposición.1,2
Se reporta una incidencia que oscila entre 0,34-2,7 casos por 100 000 al año. La EL se considera la forma de esclerodermia más frecuente en la edad infantil. La enfermedad tiene un predominio por el sexo femenino y su pico de frecuencia de aparición en la edad infantil es definido entre los 6-9 años de edad, pero existen reportes en las edades extremas de la vida.3
Se describe que el subtipo más frecuente de EL en la población pediátrica es la esclerodermia lineal (41,8-66,7 % del total de casos), le sigue la morfea circunscrita (15-37 %), la mixta (3-23 %) y la esclerodermia generalizada que se presenta entre el 6,6 y 11 % de los casos. El subtipo de esclerodermia localizada conocido como morfea panesclerótica es muy rara, poco frecuente, pero muy agresiva.3,4
Las manifestaciones clínicas de la EL suelen clasificarse en cutáneas y extracutáneas. Las lesiones cutáneas tienen una fase inflamatoria con placas eritematosas o violáceas con textura de piel normal. En estadios más avanzados aparece fibrosis progresiva y las lesiones se vuelven induradas, con un área central de color blanco-amarillento o nacarado, brillante y con un margen eritematoso o violáceo llamado lilac ring. Más adelante aparecen cambios posinflamatorios, de pigmentación, atróficos, pérdida de tejido subcutáneo y adelgazamiento progresivo de la piel. Las lesiones de la cabeza pueden acompañarse de alopecia en el cuero cabelludo o en los anejos y puede asociarse atrofia subcutánea.5,6
Las manifestaciones extracutáneas suelen presentarse entre el 20-40 % de los casos. Incluyen afectación musculoesquelética (artralgias y artritis no erosiva), afectación neurológica, gastrointestinal, cardiaca, renal, pulmonar, ocular y odontológica. También puede aparecer manifestaciones vascular, debido al fenómeno de Raynaud.5,6,7
El diagnóstico se basa en la identificación de las manifestaciones clínicas y suele corroborarse con el estudio anatomopatológico. No existen esquemas terapéuticos establecidos, pero el uso de glucocorticoides e inmunosupresores, unido al tratamiento sintomático, constituyen las principales herramientas terapéuticas de la EL.
En Ecuador, son muy escasos los reportes acerca de EL, por lo que es necesario destacar los elementos clínicos y de laboratorio que facilitan el diagnóstico temprano de la enfermedad para poder comenzar con tratamiento y minimizar el riesgo de aparición de complicaciones. Teniendo en cuenta lo infrecuente de la enfermedad y la escasez de reportes en el contexto ecuatoriano, se decide realizar el presente reporte de caso clínico en el cual se diagnostica EL en una escolar de 10 años de edad. Se considera un reporte válido para la comunidad médica.
Caso clínico
Escolar femenina, de 10 años de edad, con antecedentes de buena salud, que acudió a consulta acompañada de su madre que refirió que desde hacía alrededor de 3 años comenzó a notar la presencia de una lesión hiperpigmentada en la espalda de alrededor de 7 cm de extensión. En los últimos 6 meses la lesión ha ido aumentando paulatinamente llegando hasta alrededor de 5 cm en el momento de asistir a consulta. Adicionalmente existían antecedentes de artralgias en rodillas y muñecas sin signos inflamatorios y de intensidad moderada con escala visual de dolor en 6 puntos. Se recoge también la presencia de cefalea universal de frecuencia e intensidad variable.
Como datos positivos al examen físico se identificó la presencia de una lesión hiperpigmentada localizada a nivel de la espalda, de 7 cm de extensión, de bordes bien definidos, no elevada (Fig.). En el sistema osteomioarticular se identificó dolor a la movilización de ambos tobillos y rodillas sin signos inflamatorios.
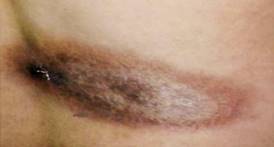
Con los datos obtenidos al interrogatorio y examen físico se procedió a interconsultar el caso con el Servicio de Dermatología que sugirieron realizar una biopsia de piel. Los resultados de la biopsia de piel informan lesión inflamatoria con presencia de infiltrado perivascular y perianexial con predominio de linfocitos y presencia de plasmáticas. Este informe confirmó el diagnóstico de la enfermedad: EL del tipo esclerodermia lineal.
A pesar de existir artralgias en rodillas y tobillos, sin signos inflamatorios y presencia de manifestaciones generales se obtuvo negatividad de los anticuerpos antinucleares, considerándose el caso como una EL. A pesar de esto, por la presencia de las manifestaciones descritas se procedió a plantear, como esquema terapéutico, la combinación de tratamiento tópico con el uso de inmunosupresores y glucocorticoides para aprovechar su acción antinflamatoria.
Desde el punto de vista tópico se procedió a utilizar clobetazol en ungüento al 1 %, que fue combinado con dosis diaria de 10 mg de prednisona y 10 mg semanales de metotrexate, calculados a dosis de 15 mg/m2 de superficie corporal. En la actualidad la paciente se mantiene en régimen de seguimiento multidisciplinario con la participación de las especialidades de dermatología, reumatología y pediatría.
Discusión
La EL es una enfermedad con un bajo porcentaje de presentación. Sin embargo, cuando se diagnostica un paciente con EL se le debe brindar un seguimiento estricto, ya que puede comenzar como un proceso local, pero también puede ser el comienzo de una enfermedad sistémica como la esclerosis sistémica.7
En el caso que se presenta es una adolescente de 10 años de edad que llevaba alrededor de 3 años con una lesión dermatológica que había ido aumentando de tamaño en los últimos meses. Estos elementos coinciden con los reportados en la literatura que expone un predominio de incidencia entre los 6 y 9 años y por el sexo femenino; claro está, estos datos no descartan su presentación en pacientes masculinos o en aquellos con un rango de edad diferente al reportado como predominante.3
El elemento clínico fundamental de esta enfermedad es la presencia de la afectación dermatológica; esta puede ser desde una afectación superficial, hasta una mayor expansión del daño llegando a afectar capas más profundas con mayor riesgo de atrofia y retracción cutánea. Este elemento, su localización y extensión es fundamental para definir la forma clínica y la gravedad de la enfermedad. La gravedad condiciona los grupos farmacológicos que se van a incluir en el esquema terapéutico.8
A pesar de ser la EL una afectación que se enfoca en la presencia de daño en la piel, pueden existir distintos tipos de afectación en otros órganos o sistemas de órganos del cuerpo humano. En el caso que se presenta se identificaron artralgias en los miembros inferiores y cefalea como expresión de compromiso neurológico. La identificación de estos elementos constituye un punto fundamental a la hora de definir el tipo de forma clínica de la EL presente en cada paciente y el esquema terapéutico a utilizar.9
Aunque se describe que el diagnóstico debe enfocarse en el elemento clínico, la realización de estudios anatomopatológicos es un factor de gran ayuda. Actúa como una herramienta que aporta elementos histopatológicos, cuya interpretación no solo confirma el diagnóstico, sino que también ayuda a comprender el mecanismo patogénico de la enfermedad. Adicionalmente puede aportar elementos importantes para interpretar la intensidad y profundidad del aumento de colágeno en las distintas capas de la piel.4,7
El tratamiento prescrito se basa en la administración de tópicos locales como clobetazol, glucocorticoides y del methotrexate. Este último es el fármaco modificador de la enfermedad de primera línea en el tratamiento de la EL, sobre todo en los subtipos que muestran afectación moderada, grave o de localización más profunda que la epidermis.3 En caso de respuesta inadecuada o intolerancia al uso de methotrexate se puede probar con otros FAME, inmunosupresores o terapia biológica.10,11
La EL es una enfermedad que puede permanecer de esa forma, pero también puede evolucionar hacia una esclerosis sistémica; con compromiso articular y extraarticular generando gran número de manifestaciones clínicas y complicaciones de la enfermedad que suponen distintos grados de discapacidad y afectación de la percepción de la calidad de vida relacionada con la salud.
Conclusiones
La esclerodermia localizada es una enfermedad rara cuyo diagnóstico se dificulta en ocasiones y se retrasa, lo que favorece las complicaciones resultantes del proceso inflamatorio mantenido. Se presenta en edades tempranas de la vida, con manifestaciones articulares y extraarticulares que diferencian cada una de sus formas clínicas de presentación. El diagnóstico es diferenciado para cada forma clínica e incluye elementos clínicos y de laboratorio. Se recomienda el uso de esteroides y methotrexate como FAME como la combinación terapéutica de elección en la mayoría de las formas clínicas de presentación.

