










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
El síndrome POEMS, definido por sus siglas como polirradiculopatía, organomegalia, endocrinopatía, proteínas monoclonales (gammapatía monoclonal), skin (alteraciones cutáneas), es un trastorno paraneoplásico, poco frecuente, crónico y limitante, caracterizado por neuropatía periférica desmielinizante y proliferación clonal de células plasmáticas. Compuesto por múltiples manifestaciones clínicas como las mencionadas en su acrónimo y que fueron descritas por primera vez por Bardwick en 1980,1,2 así como por otras, las cuales involucran diferentes sistemas como el cardiorrespiratorio, hematológico, endocrinológico, óseo y reticulonodular, puede producir confusiones y retrasos en su diagnóstico y tratamiento.1
El diagnóstico se basa en criterios clínicos y paraclínicos que requiere la confirmación de dos criterios mandatorios: presencia de polineuropatía usualmente desmielinizante y trastorno monoclonal de células plasmáticas. Además es necesario por lo menos uno de los otros criterios mayores que incluyen lesiones osteoscleróticas, factor de crecimiento endotelial vascular elevado (VEGF) o enfermedad de Castleman y por lo menos 1 criterio menor adicional, entre los cuales se puede presentar acropaquía, pérdida de peso, hiperhidrosis, hipertensión pulmonar, enfermedad pulmonar restrictiva, diarrea, vitamina B12 baja y diátesis trombótica.3
El curso de esta enfermedad suele ser progresivo, y la supervivencia general a los 10 años es variable entre los pacientes, por lo que las tasas de sobrevida pueden variar entre 55-79 % según lo reportado antes y después de 2003, respectivamente,4 de acuerdo con factores pronósticos como la presencia de hipertensión pulmonar, capacidad de difusión del monóxido de carbono (DLCO) disminuida, mayor edad, acropaquía, sobrecarga de volumen y tasa de filtración glomerular < 30 mL/min/1,73 m2.4,5,6,7 Su tratamiento se asemeja al de otros trastornos de células plasmáticas como el mieloma múltiple y la amiloidosis de cadena ligera y las recomendaciones se basan principalmente en series de casos y anécdotas.1 Múltiples avances se han realizado en cuanto al tratamiento, en el cual la gammapatía monoclonal se ha convertido en el principal objetivo a la hora de tratar este trastorno, por tanto, diferentes fármacos citotóxicos, corticoides, entre otros, tienen un papel fundamental en el tratamiento farmacológico.3,4
Actualmente, se han descrito pocos casos a nivel mundial, incluso se han reportado presentaciones atípicas, lo cual genera incertidumbre en la comunidad médica acerca de su diagnóstico y tratamiento. Por ello, el acceso a biomarcadores, imagenología y diferentes especialidades médicas, es primordial a la hora de identificar pacientes en los cuales se sospeche este síndrome.3
El propósito de este estudio es presentar dos casos de pacientes que acudieron al servicio de urgencias con manifestaciones diversas y tras la evaluación y el estudio detallado y multidisciplinario, tuvieron diagnóstico de síndrome POEMS.
Presentación de los casos
Caso 1
Paciente masculino de 72 años quien consultó al servicio de urgencias por un cuadro clínico de 4 meses de evolución consistente en pérdida de peso no cuantificada, involuntaria, asociada a hiporexia, astenia y adinamia. Un mes y medio previo al ingreso presentó disminución de la fuerza muscular proximal, bilateral, simétrica, inicialmente en hombros y antebrazos, artralgias en hombros y manos con limitación para la movilidad, sin rigidez articular, edema, calor ni rubor.
Concomitantemente, refería dolor urente simétrico en el cuello, los hombros, la cintura escapular y en ambos miembros inferiores, exacerbado en las noches, sin atenuación con analgésicos comunes. En el último mes, asoció debilidad progresiva, bilateral, simétrica, en los miembros inferiores sin limitación en la marcha.
Dos semanas antes de consultar, apareció un dolor abdominal sordo, de moderada intensidad, generalizado, asociado a distensión abdominal, con aumento del perímetro abdominal, plenitud posprandial y edema progresivo en los miembros inferiores; sin otros síntomas referidos.
El paciente tenía antecedes de hiperplasia prostática benigna, era usuario de sonda vesical permanente, padecía enfermedad pulmonar obstructiva crónica (EPOC) no estadificada y 3 meses antes de su ingreso había presentado una hemorragia de vías digestivas debido a una úlcera gástrica Forrest Ib anemizante, que fue tratada mediante escleroterapia endoscópica. También refería consumo de tabaco activo. No tenía antecedentes familiares de importancia.
Al examen físico de ingreso se encontró al paciente pálido, caquéctico, con signos vitales: tensión arterial (TA) 120/70 mmHg, frecuencia cardiaca (FC) 75 latidos/min, frecuencia respiratoria (FR) 20 respiraciones/min, temperatura 36°, saturación oxígeno (SaO2) 95 %, FiO2 21 % al aire ambiente, peso 50 kg, talla 1,61 m, índice de masa corporal (IMC) 19,3, cuello sin ingurgitación yugular, abdomen distendido, con onda ascítica positiva, extremidades simétricas con dolor a la palpación de ambos hombros, manos sin dolor articular, test de Squeeze negativo, sin artritis en codos, rodillas y tobillos, acropaquía, edema de miembros inferiores grado I fóvea positiva, perfusión distal menor de 2 s (Tabla 1).
Al examen neurológico se halló debilidad proximal 1/5 con limitación marcada para elevar ambos hombros, fuerza distal 4/5, ROT +/++++ en las cuatro extremidades, fuerza de miembros inferiores 4/5, hipotrofia muscular generalizada, normotónicas, sin déficit ni nivel sensitivo, sin signos de Hoffmann ni Tromner, respuesta plantar flexora bilateral. No tenía úlceras bucales ni nasales, alopecia, fragilidad capilar, eritema malar, sinovitis ni lesiones en la piel. El examen cardiopulmonar resultó normal.
Tabla 1 - Estudios paraclínicos realizados a los pacientes
| Leucocitos | 15,09 K/µL | 5,3 K/µL |
| Neutrófilos | 13,0 K/µL | 56 % |
| Linfocitos | 2,0 K/µL | 32 % |
| Monocitos | 0,1 K/µL | - |
| Eosinófilos | 0 K/µL | - |
| Hemoglobina | 4,80 g/dL | 10,6 g/dL |
| Hematocrito | 14,40 % | 32 % |
| VCM | 84,70 fL | - |
| MCH | 28,20 pg | - |
| RDW | 17,8 % | - |
| Plaquetas | 392 000 | 233 000 |
| LDH | 140 U/L | - |
| Creatinina | 1,14 mg/dL | 1,59 mg/dL |
| BUN | 55,3 mg/dL | 52 mg/dL |
| Sodio | 132 mmol/L | 141 mmol/L |
| Potasio | 4,9 mmol/L | 3,74 mmol/L |
| Calcio iónico | 1,00 mmol/L | 1,18 mmol/L |
| AST | 42 U/L | 5,0 U/L |
| ALT | 31 U/L | 10 U/L |
| Fosfatasa alcalina | 169,0 U/L | 71 U/L |
| CPK total | 20,00 U/L | - |
| Bilirrubina total | 0,22 mg/dL | 1,28 mg/dL |
| Bilirrubina directa | 0,21 mg/dL | - |
| Bilirrubina indirecta | 0,01 mg/dL | - |
| GGT | 96 U/L | - |
| Albúmina sérica | 1,9 g/dL | 3,2 mg/dL |
| Proteínas totales | 5,7 g/dL | - |
| PCR | 3,4 mg/L | - |
| VSG | 115 mm/h | - |
| TTP | - | 37 s |
| TP | - | 12,2 s |
Leyenda: VCM: volumen corpuscular medio; MCH: hemoglobina corpuscular media; RDW: amplitud de distribución eritrocitaria; LDH: lactato deshidrogenasa; BUN: nitrógeno ureico en sangre; AST: aspartato aminotransferasa; ALT: alanina aminotransferasa; CPK: creatinfosfoquinasa; GGT: gamaglutamil transferasa; PCR: proteína C reactiva; VSG: velocidad de sedimentación globular; TTP: tiempo de tromboplastina parcial; TP: tiempo de protrombina.
Inicialmente por el dolor abdominal, se realizaron estudios de extensión como ecografía abdominal total, que reportó hepatomegalia y ascitis en moderada cantidad, estudio del líquido peritoneal el cual evidenció un GASA de 0,97 indicativo de ascitis no hipertensiva que fue corroborada mediante estudio Doppler portal negativo. Se descartó infección primaria y secundaria del líquido ascítico. Los estudios de laboratorio están descritos en las tablas 1 y 2.
Tabla 2 - Estudios hormonales, reumatológicos y hematológicos de los pacientes
| Hormonales | - |
TSH 11,48 UI/mL T4 libre 0,52 ng/dL T3 libre 1,97 pg/mL Prolactina 101 ng/dL LH 1,4 mUI /mL FSH 1,34 mUI /mL PTH 45,6 pg/mL Cortisol 298 ug /dL Glicemia 97 mg/dL |
| Reumatológicos | - |
Complemento: Normal Panel AF: Negativo Anti-CCP: 339 Unidades ANAS y ENAS: Negativo |
| Hematológicos |
Cadenas livianas kappa en suero 5,66 g/L Vr:1,38-3,75 Cadenas livianas lambda en suero 3.29 g/L Vr: 0,93-2,42 Relación cadenas kappa/lambda en suero 1,72 Vr:0,26-1,65 Aspirado de médula ósea: negativo para infiltración neoplásica, médula ósea dentro de los parámetros normales Electroforesis de proteínas: Banda monoclonal en la región gammaglobulinas. Pico monoclonal gamma de 1.4 g/dL |
Proteínas totales en suero: 5,5 g/d Relación albúmina/globulina: 0,52 |
Durante la observación, el paciente tuvo mejoría del dolor abdominal. Sin embargo, persistió con debilidad de las extremidades superiores e inferiores y dolor asociado. Por tanto, fue valorado por el servicio de neurología clínica quien corroboró los síntomas mediante los hallazgos al examen neurológico, que evidenciaron reducción marcada de la fuerza proximal en los miembros superiores, moderada en los miembros inferiores, hiporreflexia generalizada y atrofia muscular.
Estos hallazgos permitieron localizar la lesión a nivel del sistema nervioso periférico y dentro del diagnóstico diferencial, se descartó un posible origen miopático con CPK total, aldolasa y enzimas hepáticas normales. No se consideró enfermedad de la unión neuromuscular, ya que los síntomas estaban presentes y eran constantes durante todo el día y no había tampoco exposición a tóxicos como organofosforados. Se centró el enfoque en enfermedades que comprometieran el nervio periférico, por tanto, por historia clínica se descartó la causa tóxica y enfermedad del paciente crítico.
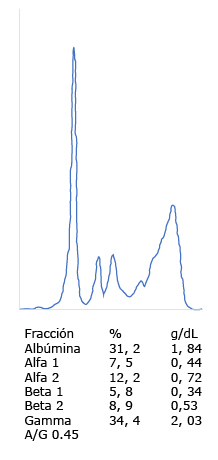
Se hicieron estudios paraclínicos que descartaron causa urémica, carencial por deficiencia de vitamina B12, infecciosa y diabética. Por la edad y los síntomas constitucionales adicionales, se hizo electroforesis de proteínas que documentó un pico monoclonal en la región gamma e inmunofijación que confirmó la elevación de las cadenas ligeras kappa y lambda en el suero y la orina (Fig. 1).
La neuropatía periférica se confirmó mediante electromiografía más neuroconducción de las cuatro extremidades con reporte de polineuropatía desmielinizante motora y sensitiva. Se inició tratamiento con el neuromodulador pregabalina a 75 mg/día.
Teniendo en cuenta el diagnóstico de polineuropatía desmielinizante, se descartó la polineuropatía desmielinizante idiopática crónica (CIDP), puesto que uno de los criterios diagnósticos de esta última implica descartar otras causas de polineuropatía desmielinizante. Por tanto, ante el diagnóstico de gammapatía monoclonal documentada, se centraron las posibilidades diagnósticas en descartar mieloma múltiple, amiloidosis del tipo AL con compromiso multisistémico y síndrome POEMS.
Se realizó estudio radiográfico de huesos largos con el fin de documentar lesiones óseas, pero fue negativo y fue complementado mediante gammagrafía ósea en la cual se describieron múltiples lesiones osteoscleróticas con compromiso de húmeros, columna vertebral y huesos iliacos (Fig. 2). Se realizó aspirado de médula ósea (AMO) el cual fue normal.
Con estos hallazgos, se descartó mieloma múltiple puesto que las lesiones en esta enfermedad suelen ser de origen lítico. El paciente no presentó compromiso renal y no se documentaron células plasmáticas en el AMO. También se descartó amiloidosis AL, puesto que en esta enfermedad es raro el compromiso óseo y la polineuropatía tiene un componente predominantemente axonal y autónomo.

Por último, se consideró el síndrome POEMS como diagnóstico principal en el paciente, ya que cumplía los dos criterios mandatorios de polineuropatía y gammapatía monoclonal, un criterio de los otros mayores (presencia de lesiones óseas osteoscleróticas) y tres de los criterios menores (organomegalia, sobrecarga de volumen extravascular, trombocitosis y el hallazgo adicional de acropaquía).
El paciente fue remitido a un centro oncológico e inició quimioterapia con melfalan-dexametasona debido al compromiso óseo extenso. Después de tres ciclos de quimioterapia, el paciente falleció por complicaciones infecciosas.
Caso 2
Paciente femenina de 46 años quien consultó por un cuadro clínico de 3 días de evolución, consistente en disnea progresiva asociada a dolor torácico de características pleurales, tos con expectoración, astenia, adinamia y edema de miembros inferiores, sin otros síntomas.
A la revisión por sistemas, refería disminución de la fuerza en los miembros superiores e inferiores de instauración progresiva durante los últimos 3 años, así como parestesias en guante y bota, marcha inestable, pérdida de peso de 8 kg en el último año y disminución del vello púbico y axilar.
La paciente tenía antecedente de artritis reumatoide e insuficiencia renal crónica; además padecía hipotiroidismo, gammapatía monoclonal, CIDP y trombosis portal con documentación de déficit de proteína S, todos diagnosticados en una hospitalización previa 3 años antes del nuevo ingreso. Era fumadora de 1-2 cigarrillos al día desde los 20 años.
Tuvo tratamiento farmacológico con rituximab un ciclo (4 dosis) 2 años antes del ingreso, metotrexate 15 mg/semanal hasta el año previo, levotiroxina 50 µg/día, enoxaparina 40 mg/día por vía subcutánea y pregabalina 150 mg/día.
Al examen físico la paciente estaba en condiciones generales regulares, con palidez mucocutánea generalizada, hipotensa (TA 70/50 mmHg), FC 90 latidos/min, FR 20 respiraciones/min, SaO2 91 % ambiente. A la auscultación cardiopulmonar se halló disminución del murmullo vesicular bibasal, estertores finos izquierdos, abdomen distendido debido a ascitis, hernia umbilical reductible, esplenomegalia dolorosa 5 cm por debajo de la reja costal izquierda, extremidades con edema grado II en los miembros inferiores, pulsos periféricos positivos, llenado capilar 3 s, piel con hiperpigmentación generalizada, engrosamiento cutáneo en antebrazos y esclerodactilia. Al examen neurológico se encontró fuerza muscular 3/5 en las cuatro extremidades, ROT: ++/+++ rotuliano bilateral e hipoestesia en el tercio distal de los miembros superiores e inferiores.
Dado que la paciente ingresó por un síndrome edematoso, dolor torácico y disnea, se solicitaron imágenes de tórax que evidenciaron derrame pleural bilateral, signos de hipertensión pulmonar precapilar y engrosamiento de los septos interlobulillares y peribronquial bilateralmente; el ecocardiograma confirmó la hipertensión pulmonar y descartó trastornos de la contractilidad que sugirieran síndrome coronario agudo o signos de insuficiencia cardiaca.
Por la presencia de ascitis, se realizó el estudio de líquido ascítico que descartó tuberculosis peritoneal. En las imágenes abdominales, se documentó esplenomegalia y cambios de micronodularidad hepática. Mediante la endoscopía de las vías digestivas altas se identificó várices esofágicas sin signos de sangrado y gastropatía por hipertensión portal, por lo tanto, ante la sospecha de cirrosis hepática, se tomó una biopsia por laparoscopía que reportó hepatitis reactiva. Los exámenes de laboratorios realizados se muestran en las tablas 1 y 2.
Se confirmó una banda monoclonal en la región gamma de 1,4 g/dL en la electroforesis de proteínas. Debido a la existencia de poliserositis, trombofilia en estudio y los síntomas neurológicos periféricos con el diagnóstico previo de CIDP, se realizó nuevamente una electromiografía con neuroconducción de las cuatro extremidades mediante la cual se evidenció polineuropatía desmielinizante crónica, con compromiso segmentario, sensitivo-motor con predominio sensitivo grave.
Llamaba la atención los signos de hipogonadismo en la paciente, por lo cual se solicitaron estudios de bioquímica hormonal, que corroboraron el diagnóstico de hipogonadismo hipogonadotrófico e hiperprolactinemia (Tabla 2).
El caso fue llevado a junta médica interdisciplinaria, conformada por los servicios de neurología, reumatología, medicina interna, neumología y hematología, quienes definieron que se trataba de un síndrome POEMS, ya que cumplía los dos criterios obligatorios de gammapatía monoclonal y polineuropatía desmielinizante, además de poliserositis, organomegalia, hipertensión pulmonar, hiperpigmentación y endocrinopatía dada por hipotiroidismo, hipogonadismo hipogonadotrófico e hiperprolactinemia de reciente diagnóstico.
Discusión
El síndrome POEMS es un trastorno paraneoplásico poco frecuente, que se presenta principalmente en la sexta década de la vida, con una ligera preponderancia masculina,5,6) caracterizado por el compromiso multisistémico que abarca diversas y heterogéneas manifestaciones clínicas. Su diagnóstico requiere un alto índice de sospecha y se sustenta en criterios clínicos y paraclínicos,1 acompañados de la presencia de gammapatía monoclonal y polineuropatía predominantemente desmielinizante. Esta última es el síntoma inicial predominante del síndrome y se caracteriza por ser subaguda, distal, simétrica, sensitivo-motora, con frecuencia muy dolorosa y asociada a alodinia e hiperpatía.8,9 Ambos casos se diferenciaron de lo descrito por no iniciar con tal cuadro clínico, sino por dolor abdominal y afectación pulmonar, motivos que llevaron a consultar por urgencias.
En el primer caso clínico reportado, el paciente consultó por dolor abdominal y durante estudios secundarios se documentó ascitis y hepatomegalia, sin otros hallazgos patológicos. Sin embargo, el síntoma que llamó la atención y motivó estudios adicionales fue la debilidad muscular y los hallazgos neurológicos como el dolor neuropático, la hiporreflexia y atrofia muscular.
En el segundo caso clínico reportado, la paciente consultó por dificultad respiratoria y síndrome edematoso, con antecedentes de afectación sistémica y de debilidad muscular progresiva asociada a trastornos hormonales, que resultaron ser un cuadro clínico poco frecuente en la literatura. Estos síntomas, junto a los hallazgos neurológicos como las parestesias en guante y en bota, orientaron a la realización de exámenes complementarios.
El descubrimiento de polineuropatía desmielinizante en ambas situaciones permitió una aproximación diagnóstica del caso, puesto que se descartaron sus causas comunes para proceder al estudio de causas infrecuentes. El principal diagnóstico diferencial es la polineuropatía crónica inflamatoria desmielinizante,10 en la cual la disociación albúmino-citológica en el estudio de líquido cefalorraquídeo permite hacer un acercamiento diagnóstico.
Es fundamental, una vez que se tiene la sospecha diagnóstica del síndrome, iniciar la búsqueda activa de las otras características clínicas que complementen los criterios diagnósticos y permitan hacer un abordaje diferencial amplio.
En caso de tener a un paciente con diagnóstico de neuropatía y cualquiera de los siguientes factores, se debe realizar una búsqueda activa del síndrome POEMS a través de la identificación de proteína monoclonal, trombocitosis, anasarca o papiledema, así como pacientes con diagnóstico de polineuropatía desmielinizante inflamatoria crónica que no responden a la terapia estándar para dicha enfermedad.2
Nuestros pacientes se atendieron a partir del diagnóstico neurofisiológico y electroforético, así como de un amplio estudio de laboratorio e imágenes. Se debe resaltar que la gammagrafía ósea es un examen clave, puesto que además de evidenciar las lesiones osteoscleróticas permite caracterizarlas; esto último es fundamental para descartar otros trastornos de células plasmáticas como la amiloidosis AL y el mieloma múltiple, lo que concuerda con lo expuesto en la literatura.
Finalmente se lograron integrar otros hallazgos clínicos y paraclínicos, que fueron fundamentales para configurar el diagnóstico final del síndrome POEMS. Estos resultaron por un lado ser el primer caso reportado con hipogonadismo y por otro se convirtieron en los primeros casos del síndrome POEMS reportados en Colombia.
Conclusiones
El síndrome POEMS es un trastorno paraneoplásico raro y poco frecuente que requiere alto índice de sospecha clínica por su amplia variabilidad de síntomas y que se puede presentar con cuadros no reportados aún en la literatura, como alteraciones hormonales e hipogonadismo. Nuestra intención fue motivar a los distintos clínicos a unir las piezas del rompecabezas para lograr el acercamiento diagnóstico de una enfermedad rara y desconocida por muchos.

