










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkCorSalud
versión On-line ISSN 2078-7170
CorSalud vol.10 no.1 Santa Clara ene.-mar. 2018
ARTÍCULO DE REVISIÓN
Ventrículo izquierdo no compacto: panorámica y arritmogenia
Left ventricular non-compaction cardiomyopathy: outlook and cardiac arrhythmias
Margarita Dorantes Sánchez, Ana M. Jerez Castro, Sheila Hechavarría Poymiró
Servicio de Arritmias y Estimulación Eléctrica Cardíaca. Instituto de Cardiología y Cirugía Cardiovascular. La Habana, Cuba. Correo electrónico: dorantes@infomed.sld.cu
RESUMEN
Las miocardiopatías constituyen un grupo importante y heterogéneo de enfermedades del miocardio asociadas a disfunción mecánica, eléctrica, o ambas. El ventrículo izquierdo no compacto es una miocardiopatía familiar de etiología incierta de la que se desconocen sus exactas incidencia y prevalencia. Se caracteriza por un aumento en la masa trabecular del VI en contraste con una fina capa epicárdica compacta que puede visualizarse con técnicas de imagen que confirman el diagnóstico. En este artículo se describen la clasificación de MOGE(S) para las miocardiopatías, los trastornos electrocardiográficos que pueden encontrarse en pacientes con ventrículo izquierdo no compacto, el papel de la estimulación eléctrica programada del corazón y otros aspectos de interés de esta enfermedad. Además, se presentan algunos trastornos electrocardiográficos demostrativos (criterios de Stollberger y Jenni) encontrados en pacientes afectados.
Palabras clave: ventrículo izquierdo no compacto, miocardiopatía espongiforme, arritmias cardíacas, miocardiopatías, clasificación.
ABSTRACT
Cardiomyopathies are an important and diverse group of myocardial diseases associated with mechanical, electrical, or both dysfunctions. The left ventricular (LV) non-compaction is a familial cardiomyopathy of uncertain etiology, whose exact incidence and prevalence are unknown. It is characterized by an increase in the trabecular mass of the LV in contrast to a thin compact epicardial layer that can be visualized with imaging techniques that confirm the diagnosis. In this article is described the classification of MOGE (S) for cardiomyopathies, electrocardiographic disorders that can be found in patients with left ventricular non-compaction, the role of programmed electrical stimulation of the heart and other aspects of interest of this disease. In addition, some demonstrative electrocardiographic disorders (Stollberger and Jenni criteria) found in affected patients are presented.
Keywords: left ventricular noncompaction, spongiform cardiomyopathy, cardiac arrhythmias, classification.
LAS MIOCARDIOPATÍAS (CARDIOMYOPATHIES)
Las miocardiopatías (MCP) constituyen un grupo importante y heterogéneo de enfermedades del miocardio asociadas a disfunción mecánica (diastólica o sistólica), eléctrica, o ambas, que usualmente exhiben hipertrofia ventricular inapropiada o dilatación. Sus causas son variadas, con frecuencia genéticas. Se encuentran confinadas al corazón o como parte de alteraciones sistémicas generalizadas, y pueden conducir a la muerte cardiovascular o a insuficiencia cardíaca (IC) progresiva1,2
Importancia de las clasificaciones
Los sistemas de clasificación aparecen en todos los aspectos de la vida, aunque para ser útiles deben cambiar al ritmo de los nuevos conocimientos3. Una clasificación sirve como un puente entre la ignorancia y el conocimiento4.
En cuanto a las MCP, ha llegado el momento de nuevas definiciones y clasificaciones, de redefiniciones y reclasificaciones. Existe una verdadera revolución en el campo de estas complejas y heterogéneas enfermedades. En 1980 se elaboró su clasificación por la Organización Mundial de la Salud y la International Society and Federation of Cardiology, y en 1995, la primera, clasificó las enfermedades miocárdicas con disfunción cardíaca, no sólo con depresión de la contractilidad y mala función diastólica sino que incorporó los trastornos del ritmo, y se basaba en la genética molecular y en la genómica, con inclusión del ventrículo izquierdo no compacto (VINC) entre las no clasificadas5.
¿POR QUÉ NUEVAS CLASIFICACIONES? SU EVOLUCIÓN?
Qué es el ventrículo izquierdo no compacto: Su lugar dentro de las miocardiopatías
Las MCP se han clasificado como primarias o secundarias, según criterios morfológicos y funcionales. La American Heart Association clasificó el VINC (también denominado miocardio espongiforme, miocardio fetal, miocardio ventricular izquierdo no compacto, síndrome de hipertrabeculación, MCP no compacta, MCP espongiforme, hipertrabeculación del ventrículo izquierdo), como una categoría diferente (2006) y se incluye dentro de las MCP primarias, genéticas o no, con afectación cardíaca única o predominante, enfermedades cardíacas hereditarias con riesgo de arritmias ventriculares, bloqueo aurículo-ventricular completo y episodios de muerte súbita (MS), con alteraciones en las proteínas miocárdicas y apariencia morfológica distintiva (esponjosa) del miocardio ventricular izquierdo, con excesivas y profundas trabeculaciones que interesan más a las porciones distal apical y mediolateral inferior del miocardio del ventrículo izquierdo (VI), con profundos recesos intertrabeculares (sinusoides) en comunicación con la cavidad ventricular y que resulta de un defecto en la embriogénesis normal. Puede cursar aislado o con otras cardiopatías congénitas, ser familiar o no y solaparse con la MCP hipertrófica (H), la dilatada (D) o la restrictiva, lo que sugiere una enfermedad continua asociada a mutaciones genéticas de las sarcómeras, y a cardiopatías congénitas y enfermedades sistémicas neuromusculares y metabólicas1,6-9. El VINC se contempla como una enfermedad rara, congénita, genética primaria, que resulta de un defecto intrauterino y de una anormalidad del proceso de compactación miocárdica. Existen profundos recesos intertrabeculares en segmentos intensamente hipertrofiados y con frecuencia hipocinéticos del miocardio del VI; puede asociarse a IC, eventos embólicos, arritmias y MS cardíaca9. Su diagnóstico se realiza sobre todo por ecocardiograma y por resonancia magnética nuclear10.
El VINC es una MCP familiar (su exacta incidencia y prevalencia se desconocen), de etiología incierta para la que se ha propuesto un origen genético6,7. Desde el punto de vista anatomopatológico se caracteriza por un aumento en la masa trabecular del VI (no compacto) en contraste con una fina capa epicárdica compacta que puede visualizarse con técnicas de imagen que confirman el diagnóstico10,11.
Normalmente el VI es liso en su interior con excepción de los dos músculos papilares (anterior o lateral, y posterior o medial), donde se anclan las cuerdas tendinosas de la válvula mitral. No existen trabéculas musculares que permitan dividir la cavidad en diferentes porciones como en el ventrículo derecho2.
En el VINC el miocardio puede tener una función sistólica o diastólica normal, o no, cursar con dilatación ventricular o hipertrofia, y su talla y función pueden cambiar de manera inesperada (fenotipo ondulante)6. Aquellos con tratamiento crónico para lograr su compensación, a veces presentan empeoramiento de su IC de manera aguda.
Esta enfermedad se ha planteado como un síndrome, no siempre genético, otros lo conciben como una MCP genética o mixta12. En el 2006 se reconoció como una MCP genética (American Heart Association), cuya nomenclatura y patogenia se caracterizan por un engrosamiento de la pared ventricular regional y recesos trabeculares profundos. Su genética puede solaparse con fenotipos de otras MCP genéticas o mixtas (MCP-D, MCP-H y otras), con variaciones patológicas y fisiopatológicas de la arquitectura miocárdica y de su función. Por diversas causas y mecanismos fisiopatológicos, el desarrollo en la estructura miocárdica ventricular puede ser complejo y desordenado. Existe un carácter morfológico de la estructura miocárdica con un amplio espectro de variantes normales hasta los fenotipos patológicos del VINC, que reflejan la estructura embriogénica del corazón humano con una detención en el proceso de compactación durante el primer trimestre, con o sin otras malformaciones cardíacas congénitas. Se encuentra una diversidad genética compleja pero también etiológica, clínica, patológica y en el diagnóstico por imágenes, con variaciones en el desarrollo miocárdico en relación con las mutaciones genéticas y en el fenotipo de uno o de un grupo de genes, por procesos interactuantes y alteración de la modulación en la expresión genética, funcional y otras.
El primer caso de VINC fue descrito en 1926 en una autopsia de un recién nacido con una cardiopatía congénita, y por ecocardiograma en 1984; a partir de entonces su reconocimiento fue en aumento. De todos modos se discute si se trata de una MCP separada o de un carácter fenotípico morfológico compartido por distintas MCP13.
En virtud de los extraordinarios avances de la genética molecular en los últimos años, han surgido nuevas clasificaciones que intentan ser más completas, rigurosas, con apertura para los futuros avances en la era de la biología y la genética molecular de las enfermedades cardiovasculares, pero al mismo tiempo deben ser simples, claras y flexibles.
El VINC es un trastorno de la morfogénesis endomiocárdica que resulta en múltiples trabeculaciones en el miocardio del VI, la literatura sugiere que es rara en adultos y se asocia a un pobre pronóstico. Como esta alteración puede estar presente desde el nacimiento, varios estudios lo consideran enfermedad familiar, asintomática en algunos pacientes. Murphy14 plantea que hay una larga fase preclínica de la enfermedad y el pronóstico no es representativo de su verdadera historia natural.
Durante el desarrollo embrionario normal, las trabeculaciones endomiocárdicas emergen de la región apical de los ventrículos primitivos hacia el día 32 de la vida fetal, a través de un proceso de reabsorción y remodelación. El miocardio empieza como una malla laxa de fibras musculares que gradualmente se condensa de endocardio a epicardio, lo que resulta en compactación de la superficie endocárdica entre la quinta y la octava semana de la vida fetal, más completa en el VI que en el derecho. El cese en la morfogénesis endomiocárdica normal resulta en el VINC y representa un defecto del proceso de compactación13,14.
El examen histológico evidencia una continuidad entre el endocardio del VI y los recesos interventriculares profundos, adecuados para formar un sustrato que facilita la propagación de circuitos arrítmicos reentrantes15.
Durante el desarrollo embriológico normal, el corazón es una malla esponjosa de fibras musculares y trabeculaciones separadas por recesos; antes que se desarrollen los vasos coronarios los recesos intertrabeculares o sinusoides comunican con el cavum del ventrículo para recibir sangre. Luego del desarrollo coronario, el miocardio ventricular gradualmente se compacta y los recesos se transforman en capilares. La compactación trabecular ocurre entre las 12-18 semanas de gestación, de la base al ápex. En el VINC no sucede esto y se desarrolla una capa endomiocárdica engrosada no compacta con prominentes trabéculas en continuidad con la cavidad del VI y falta de comunicación con la circulación epicárdica, con profundos recesos y una capa epicárdica compacta fina, en virtud de un déficit en la regresión del miocardio no compacto16.
La orientación genómica y molecular facilita la interacción entre la clínica y la investigación, entre las complejas relaciones genotipo-fenotipo y deja ventanas abiertas para el desarrollo de la genética molecular de la enfermedad miocárdica, al tiempo que permite la inclusión de alteraciones descritas recientemente y su entrada al complejo y heterogéneo campo de estas enfermedades, que a lo largo del tiempo se han clasificado de distintas maneras. Todo ello permite que las enfermedades arrítmicas genéticas (más frecuentes de lo que se anticipaba y causa importante de MS), se integren a las MCP, crezca la lista de los manifestaciones arrítmicas hereditarias y se incluyan enfermedades descritas recientemente o mejor conocidas ahora, como las canalopatías iónicas16.
El VINC se incluye entre las MCP no clasificadas por la Organización Mundial de la Salud y la European Society of Cardiology, y como MCP genética por la American Heart Association4,16,17.
La clasificación MOGE(S) describe el fenotipo, como «banderas rojas» la afectación de otros órganos, y la causa genética o no de la enfermedad (Tabla 1)10,18.

La M se relaciona con el diagnóstico descriptivo del fenotipo, pero puede haber solapamiento de ellos, con marcadores clínicos (por ejemplo, bloqueo aurículo-ventricular, Wolff-Parkinson-White y otros), y afectación solo del corazón o no. La E ofrece la información esencial para el tipo de MCP (familia, paciente, genética).
Se trata de una nosología descriptiva que combina el carácter morfofuncional y la afectación de órgano-sistema con el patrón hereditario familiar, e identifica el defecto genético u otras causas. Resulta una revolución para los genetistas clínicos introducir el concepto diagnóstico de «banderas rojas» de Arbustini et al.10, con marcadores clínicos que pueden guiar la investigación genética a un gen específico. Es una nueva clasificación más detallada con respecto a las americanas y a las europeas previas. Puede ser útil para las MCP hereditarias pero es difícil de aplicar en la displasia arritmogénica de ventrículo derecho porque incluso la onda épsilon, considerada por muchos años marcador de la enfermedad, puede representar una «bandera roja» solo cuando está presente en el electrocardiograma y el diagnóstico de esta enfermedad requiere varias banderas de este tipo10.
Este sistema permite acercarse al problema no resuelto de la correlación fenotipo-genotipo, que considera las familias con la misma mutación, pero sólo en estos casos es posible emplear los datos genéticos para el pronóstico y la terapia de la MCP hereditaria, aunque se conoce que muchas mutaciones son únicas18,19.
Las MCP son enfermedades familiares en su mayor parte; el tamizaje familiar identifica a los asintomáticos y a los parientes con caracteres tempranos de la enfermedad. En los últimos 50 años las clasificaciones se han basado en fenotipos morfofuncionales, lo que ha permitido a los cardiólogos agrupar convenientemente las amplias categorías descriptivas. Sin embargo, el fenotipo no siempre conforma las características genéticas, puede no permitir la estratificación de riesgo y no provee el diagnóstico preclínico en los familiares. Actualmente se practican más pruebas genéticas, que ya constituyen una parte del trabajo clínico; basado en la heterogeneidad genética se acuñan nuevos nombres para describir las MCP asociadas a mutaciones de diferentes genes y se requiere una nosología comprensible que pueda informar el fenotipo clínico y el compromiso de otros órganos distintos al corazón, así como el genotipo y el modo de herencia. La reciente propuesta del sistema nosológico MOGES incluye todas estas características con el empleo de los criterios del American College of Cardiology/American Heart Association y la clase funcional de la New York Heart Association. Esta nomenclatura se auxilia con aplicaciones computacionales, ayuda a describir las MCP sintomáticas y asintomáticas y las familiares en el contexto de las pruebas genéticas; resulta flexible, permite su expansión, ayuda a entender sus bases etiológicas, a describir los complejos genéticos y da la oportunidad de crear registros más completos20.
Una posible limitación del MOGES es la falta de información en cuanto a uno de los más importantes problemas clínicos en las MCP: las arritmias. Aunque la clasificación de éstas no se encuentra entre los objetivos del MOGES, ya existen sugerencias de expandir la S con la inclusión de los trastornos del ritmo. El comité encargado de esta clasificación trabaja con los electrofisiólogos para desarrollar una descripción rápida y clínicamente útil de los trastornos del ritmo como una segunda S21.
Este sistema nosológico parece complejo y complicado en la práctica pero si se siguen los pasos progresivos, resulta aplicable y no obliga al clínico a la prueba genética. Otras ventajas son descritas en el recuadro21.
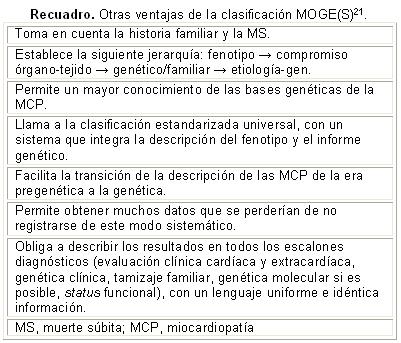
El sistema de Arbustini et al.10 es flexible y adaptable, precisa la etiología con fenotipos clínicos y, por inferencia, se adentra en el pronóstico y el tratamiento de estos pacientes. En el término genérico de MCP se comprende cualquier enfermedad del miocardio que no se explica por estrechez de las arterias coronarias o sobrecarga anormal de los ventrículos; se trata de trastornos que nacen dentro del miocardiocito o de la matriz extracelular. Los clínicos las han agrupado en subcategorías sobre la base de la morfología y de la función ventricular, con aproximación a la presentación clínica y a la estrategia terapéutica pero resultan limitadas al no considerar la etiología ni excluir los fenotipos leves o intermedios que no cumplen los criterios diagnósticos convencionales. El mencionado sistema10 intenta abarcar la complejidad fisiopatológica de las MCP, representa la progresión lógica del enfoque diagnóstico propuesto en recientes reuniones de la European Society of Cardiology, en el cual para aproximarse al diagnóstico se combinan los parámetros cardiológicos convencionales con otros no cardíacos y moleculares. Los grupos de modelos clínicos o «banderas rojas» diagnósticas pueden emplearse para identificar subtipos genéticos específicos de MCP que requieren estrategias individualizadas de atención y tratamiento, pero hay que considerar la clase morfofuncional (M) que intenta resumir las características fenotípicas y las pistas diagnósticas, tales como el bloqueo aurículo-ventricular y otras.
Existen varías críticas al sistema: si se trata de una enfermedad temprana, si no se adoptan los mismos criterios o si son datos inespecíficos. Aunque se reconoce que establece un puente entre las nuevas disciplinas (como la proteómica y la genómica) y la medicina clínica, con utilidad diagnóstica y pronóstica aún por definir3. Es necesario lograr una integración entre las ciencias básicas y la medicina clínica. Según Elliott3, Paul Wood en 1950 expresó: "...I have attempted to maintain a proper balance between man and his instruments, between experienced opinion and statistics, between traditional views and heterodox, between bed-side medicine and special tests, between the practical and the academic, and so to link the past with the present".
El VINC es una MCP primaria con un patrón morfológico específico, que tiene una estructura de 2 capas del miocardio: una epicárdica compacta delgada y una endocárdica no compacta intensamente engrosada, que consiste en una malla trabecular con profundos espacios endocárdicos que ocurren en ausencia de otras lesiones congénitas coexistentes22.
Con respecto al fenotipo y al genotipo puede encontrarse un solapamiento del VINC con otras MCP, no existe una histología característica para confirmar su diagnóstico mediante imágenes; todos los criterios diagnósticos intentan describir las particularidades de una afectación tridimensional, compleja y aleatoria, a partir de imágenes bidimensionales; no existe un patrón de referencia (gold standard) con el cual comparar estos casos y se conocen poco las características precisas de la trabeculación en un corazón normal, así como los fenotipos leves que pueden encontrarse en una enfermedad que parece familiar.
El VINC puede ser esporádico o familiar (mutaciones en las mitocondrias, el citoesqueleto y las proteínas del sarcómero). En sujetos saludables puede cumplir los criterios diagnósticos de imágenes y los niños pueden presentar síndrome de MS infantil. Como se ve en familias con MCP-H o -D, surgen dudas en cuanto a aceptarla como una enfermedad diferente. La extensión del miocardio compacto puede ser un rasgo dentro de la población y las técnicas de imagen detectar variaciones sutiles en la morfología dentro de la normalidad, no como base para causar una disfunción miocárdica. El VINC puede ser secundario a una alteración genética bien tolerada cuando el corazón es normal, pero se requieren criterios diagnósticos más restringidos. Es decir, existe la duda en cuanto a si el VINC es causa, factor contribuyente o epifenómeno en estos pacientes. En presencia de una mutación genética, la disrupción de la función del miocito a nivel molecular puede ser una enfermedad primaria determinante sin ventrículo compacto, que aparecería como una respuesta de remodelación mal adaptada que combina la enfermedad primaria con isquemia subendocárdica y fibrosis23.
El electrocardiograma y las arritmias en el ventrículo izquierdo no compacto
Estos pacientes tienen un amplio espectro clínico, desde el descubrimiento de la enfermedad en sujetos asintomáticos hasta aquellos que desarrollan arritmias ventriculares peligrosas para la vida, IC izquierda, derecha o ambas, y eventos embólicos sistémicos6,22. Los síntomas pueden ser inducidos por el ejercicio o persistir en reposo. Steffel22 estudió 78 pacientes que presentaron trastornos de la conducción intraventricular (en especial bloqueo de rama izquierda), signos de hipertrofia ventricular izquierda y anormalidades de la repolarización. Un 13% tuvo electrocardiograma normal (sujetos más jóvenes, menos IC y lesiones estructurales más leves). En general no se encuentran hallazgos electrocardiográficos específicos aunque no suele hacerse un análisis sistemático del electrocardiograma. Existe un notable solapamiento entre el bloqueo de rama izquierda en particular, el retardo de la conducción auricular, el intervalo PR largo o el bloqueo aurículo-ventricular, el aumento del intervalo QT, la reducida función sistólica del VI y la dilatación del VI y de la aurícula izquierda. Los pacientes con signos de hipertrofia ventricular izquierda tuvieron eventos embólicos sistémicos con mayor frecuencia. Hubo una mortalidad de 35% en 44 meses de seguimiento (MS cardíaca en la mitad de los casos), se presentó taquicardia en un 41% y se ha mencionado que el electrocardiograma es anormal en el 94% de los pacientes (con mayor frecuencia de bloqueo de conducción en las ramas izquierda o derecha, y repolarización anormal).
Steffel22 realizó una correlación entre el ecocardiograma y la clínica: IC, retardo de la conducción intraventricular (en especial bloqueo de rama izquierda), retardo de la conducción auricular, taquicardia/fibrilación ventriculares, asistolia y Wolff-Parkinson-White (posiblemente por interrupción del proceso normal de desarrollo del anillo fibroso). Doce pacientes con VINC recibieron cardioversor-desfibrilador automático implantable (CDAI) como prevención primaria o secundaria en casos de arritmias potencialmente letales; en alguno se practicó estimulación eléctrica programada del corazón (EEPC) para estratificar riesgo. Se encontraron trastornos de la conducción intraventricular (50%), alteraciones de la repolarización manifestada como onda T negativa en derivaciones precordiales (70%), intervalo QT largo (50%), hipertrofia del VI (30%) y trazado normal (5%)11,22.
Duru y Candinas24 informaron de un caso con taquicardias ventriculares clínicas diferentes que no se reprodujeron en la EEPC. Se han requerido como tratamiento: fármacos antiarrítmicos, anticoagulación, marcapasos, CDAI (episodios de MS), ablación, resincronización miocárdica, terapia génica y hasta trasplante por disfunción progresiva del VI11.
El VINC puede ser asintomático por años o desarrollar una MCP-D e IC, síncope, embolismo, arritmias supraventriculares y ventriculares (extrasístoles, taquicardia y fibrilación), y MS cardíaca11,14,25. Murphy et al.14 estudiaron 45 pacientes con VINC para definir el pronóstico y la incidencia familiar, y encontraron electrocardiograma anormal (91%), dilatación del VI (66%), taquicardia ventricular no sostenida (20% en estudio Holter) y tromboembolismos (4%). Apreciaron un mejor pronóstico que lo informado con anterioridad y otros de sus resultados fueron: bloqueo de rama izquierda (29%), Q patológica (9%), pobre progresión de la onda R (7%), cambios del segmento ST (9%), inversión de la onda T (16%) y fibrilación auricular. Se implantaron marcapasos en algunos casos y en otros, CDAI; ésta última como una opción terapéutica en el caso de arritmias sintomáticas o baja fracción de eyección, MS cardíaca, síncope, arritmias sostenidas y taquicardia ventricular no sostenida sincopal.
No existen muchos datos de seguimiento a largo plazo en pacientes con CDAI, tanto en la prevención primaria (sin arritmias potencialmente letales documentadas) como secundaria15. Hay una alta prevalencia de arritmias supraventriculares, fibrilación y flutter auriculares (las hubo en 8 de 12 pacientes estudiados por Kobza et al.15); taquiarritmias ventriculares (38-47%) y MS cardíaca (13-18%). La elección del CDAI se hace de acuerdo con factores clínicos, frecuencia de la taquicardia ventricular y de las supraventriculares, sintomatología, indicación concomitante de la estimulación biventricular, signos de IC, fracción de eyección baja, QRS mayor o igual a 120 ms, y dos o tres criterios adicionales de disincronía: retardo aórtico preeyección de más de 140 ms, retardo interventricular mecánico de más de 40 ms o activación retardada de la pared posterolateral del VI. En general se trata de series de pocos pacientes, sin grupo control y con seguimientos cortos.
Lo trascendente es que el VINC es un sustrato altamente arritmogénico, Yin12 habla de arritmias ventriculares en un 13% y flutter/fibrilación auriculares en un 20% de los casos.
En pacientes con VINC familiar, Murphy et al.14 encontraron que los parientes presentaron más MCP-D que VINC y se han publicado casos muy sintomáticos, con alta incidencia de arritmias ventriculares e IC progresiva14.
Los pacientes con VINC pueden tener un pronóstico más favorable que lo descrito hasta ahora pues en la forma familiar hay un espectro de anormalidades que se solapan con la MCP-D y se sugiere que estas enfermedades comparten una etiología común. Muchos pacientes del estudio de Murphy tuvieron un fenotipo leve con menor incidencia de muerte, embolia o arritmias ventriculares documentadas; lo cual puede explicarse por una selección de sujetos y de tamizaje, además de que al mejorar el diagnóstico ecocardiográfico se facilita la detección de casos asintomáticos, no reconocidos antes. El VINC se detectó retrospectivamente en sujetos diagnosticados como MCP-D, esto sugiere que su frecuencia en la población con IC puede ser infraestimada por una imagen inadecuada de los segmentos apicales del VI y aumentaría con mejores imágenes cardíacas; además, existe un período de gestación silente antes del inicio de la enfermedad clínica e incluso se discute si el VINC es un subtipo o una variante de la MCP-D más que una MCP distinta14.
Celiker9 estudió 11 niños con VINC y anormalidades del ritmo: palpitaciones, síncope, bradicardia, arritmias ventriculares, trastornos de los nodos sinoauricular y aurículo-ventricular, síndrome de Wolff-Parkinson-White (en éste, por déficit en la regresión de continuidad del desarrollo embrionario aurículo-ventricular anatómico y eléctrico).
Ikeda16 plantea que el diagnóstico del VINC se realiza con más frecuencia actualmente, pero persisten problemas diagnósticos y la conducta terapéutica se hará de acuerdo a las manifestaciones clínicas, por lo que se requieren registros multicéntricos para precisar mejor estos aspectos, además de los avances en las imágenes y los estudios genéticos. El espectro va desde los sujetos asintomáticos hasta los graves, con expresiones fenotípicas variables de otras MCP, y con una mortalidad inicial de 35-47%, muerte o trasplante (26%), taquicardias ventriculares (2-62%) y taquicardia ventricular sostenida o no sostenida en el Holter (27%).
Brescia et al.13 estudiaron a 242 niños en el período 1990-2009 e informaron: mortalidad 12,8%, anormalidades electrocardiográficas 87% (las más frecuentes, hipertrofia del VI y anormalidades de la repolarización), arritmias 33,1%, MS 6,2% y taquicardia ventricular 17,4%. Otros hallazgos fueron: inversión de la onda T, anormalidad del segmento ST, crecimiento auricular, desviación del eje a la izquierda, aumento del intervalo QT, preexcitación, taquicardia auricular y otras arritmias supraventriculares, flutter y fibrilación auriculares, y ritmo acelerado de la unión.
La arritmogenia en el VINC puede explicarse por dispersión de la repolarización, isquemia miocárdica y causas genéticas, en una población de gran heterogeneidad y con la posible existencia de subtipos (normales, dilatados, hipertróficos o mezcla de ellos), cuyo espectro se mueve desde una mortalidad alta con progresiva disfunción miocárdica, hasta bajo riesgo de MS si las dimensiones y la función cardíacas son normales.
Stollberger et al.26 estudiaron 105 pacientes (1995-2011) en los cuales el VINC se asociaba a enfermedades neuromusculares. Analizaron las anormalidades electrocardiográficas e informaron: alteraciones del intervalo ST y de la onda T, bloqueo fascicular anterior izquierdo, fibrilación auricular, ensanchamiento del QRS, ondas Q anormales, trastornos de la conducción intraventricular, hipertrofia del VI, bajo voltaje, bloqueos de ramas derecha e izquierda, PR prolongado, aumento del intervalo QT, taquicardia sinusal y, sobre todo, precisaron los cambios evolutivos de estos trastornos. Sin embargo, uno se pregunta cuánto atribuir al VINC y cuánto a los trastornos neuromusculares asociados.
Existen dudas en cuanto a la historia natural del VINC porque los estudios son pequeños, pero en todo caso recordemos que Viskin y Rogowski27 hablan del efecto fundador o descubridor, y aluden a descripciones iniciales de la MCP-H de centros terciarios que planteaban un pronóstico muy malo. Posteriormente y ante nuevos estudios comunitarios, el cuadro se presentó menos ominoso y más balanceado; más tarde se reconoció algo similar en el síndrome de Brugada. Por otro lado, los registros informan con mayor frecuencia los casos sintomáticos, los más graves y complejos, y los asintomáticos pueden escapar al diagnóstico médico. En la descripción de una enfermedad de reciente conocimiento suele sobreestimarse su morbilidad y mortalidad, con el tiempo los asintomáticos se identifican mejor y se logra una percepción más realista del problema, que es lo que se conoce como efecto descubridor27.
PAPEL DE LA ESTIMULACIÓN ELÉCTRICA PROGRAMADA DEL CORAZÓN
La EEPC puede ser útil y tener algún valor predictivo en las arritmias sintomáticas o sincopales, la distinción entre trastornos supraventriculares o ventriculares y su tratamiento, la relación con la fracción de eyección baja, la prevención de la MS cardíaca y el síncope, o ante una causa desconocida con función deprimida del VI o una enfermedad cardíaca estructural. Aunque se discute su papel para estratificar riesgo _o ni se menciona_, Kobza15 la empleó en pacientes con VINC, con un intenso protocolo de 500, 400 y 330 ms de longitud de ciclo básico y 3 extraestímulos, con intervalo mínimo de acoplamiento de 180 ms, desde el ápex del ventrículo derecho y del tracto de salida.
En nuestra opinión, en general la EEPC es útil en diversas enfermedades en las cuales se emplea, pero no dice la última palabra como se pensaba hace años; sus resultados deben considerarse a otro nivel en la actualidad, es un procedimiento con limitaciones y así debe tomársele en cuenta. Pueden suceder varias cosas (Figura 1):
- No reproducirse en el laboratorio la misma arritmia clínica.
- Originarse una artificial (de laboratorio) o una preclínica.
- Reproducir la clínica y que ella no se presente en la vida real con posterioridad o viceversa, no reproducirla y que se origine luego en la clínica.
- Tratarse de una arritmia automática, o que siendo además reentrante no se pongan de acuerdo los tres elementos del triángulo (sustrato, disparador y modulador) y, por tanto, no sea reproducible.

Debe dársele su real valor pronóstico y ser cautelosos a la hora de decidir una terapéutica (por ejemplo el CDAI), basados en el resultado de la EEPC. Por otra parte, en algunas situaciones las arritmias son malamente reproducibles en el laboratorio de electrofisiología. Debe concedérsele su verdadero valor al procedimiento, conociendo su variablidad de un momento a otro en un mismo paciente y la existencia de respuestas específicas e inespecíficas28,29. Se recomienda leer y analizar dos trabajos fundamentales de Josephson30,31 para saber qué queda para la EEPC en la era del CDAI y de la ablación, publicaciones separadas entre sí por 10 años, en las cuales se considera que la electrofisiología clínica cardíaca se encuentra en una encrucijada, carece hoy de pensamiento crítico, de entendimiento básico y está en crisis de credibilidad. Dice Josephson: "If one asks me where we go from here, I would respond: back to basics learn-electrophysiology"30,31. Es necesario e imprescindible saber qué puede esperarse de la EEPC y qué no debe esperarse de ella; en ocasiones existen modas en la práctica de algunos procedimientos y en ellos se basan las decisiones terapéuticas. Debe conocerse qué se hacía en el pasado y qué debe hacerse ahora: no sustituir la electrofisiología por la electrotecnología, que deben marchar aunadas, como mismo lo deben hacer la electrocardiografía y la electrofisiología.
A 50 años de establecerse la EEPC como procedimiento clínico, varias de sus indicaciones y conceptos han cambiado y se han precisado algunas de sus limitaciones (si bien sigue siendo una herramienta fundamental de la arritmología). La función del laboratorio clínico de electrofisiología cambió de lo diagnóstico y artístico a lo terapéutico por excelencia, aunque debe existir una absoluta conexión en ambos sentidos: el entendimiento de los sustratos arrítmicos y su ablación; no puede haber divorcio entre la electrofisiología clínica y la electrotecnología, que se enriquecen mutuamente. Se le debe conceder su verdadero lugar a la EEPC, que emplea desencadenantes en definitiva artificiales que pueden no corresponder a los clínicos y que no controla los elementos moduladores (sólo de modo limitado el sistema nervioso autónomo), y profundizar en el triángulo de toda arritmia reentrante: el sustrato arritmogénico, el elemento disparador o desencadenente y el factor modulador, para insertarse de manera integral en el proceso arrítmico, sin olvidar la posible variabilidad de un estudio electrofisiológico a otro en un mismo paciente.
El hecho de que durante tantos años se discuta y publique sobre este tema y las muchas opiniones contradictorias entre notables investigadores, indica que es un problema aún no resuelto. En un primer momento la EEPC tuvo un papel preponderante para estratificar riesgo y se le concedió la mayor confiabilidad al tomar decisiones terapéuticas, luego todo ello tomó su nivel y, sin negar su contribución, se sabe que no es algo absoluto ni siempre dice la última palabra. A lo largo del tiempo han surgido varias preguntas: ¿cuál es la verdadera utilidad de la EEPC para estratificar riesgo?, ¿cuánto pesa en la decisión terapéutica de colocar un CDAI, por ejemplo?, ¿qué valor tiene la inducibilidad de una arritmia ventricular maligna en el laboratorio para predecir el debut o la recidiva en el seguimiento de un paciente? Hay incertidumbres y limitaciones, quedan cosas por definir en cuanto a los protocolos, los registros, los falsos positivos y negativos, y los sitios de estimulación28,29,32.
Aun cuando se trata de afecciones diversas, recordemos algunos puntos muy discutidos en el VINC y en el síndrome de Brugada en cuanto a problemas conceptuales relativos a la EEPC y a otros aspectos: su incierto papel para estratificar riesgo y tomar decisiones terapéuticas, la existencia de signos y síndromes o signos que pueden evolucionar hasta hacerse síndromes, el solapamiento de otras canalopatías al síndrome de Brugada e igualmente el solapamiento de otras MCP al VINC, el amplio espectro clínico que va desde los sujetos asintomáticos hasta los de mayor gravedad, y las pistas de una enfermedad orgánica subyacente en el síndrome de Brugada28,29,33.
ALGUNAS OTRAS ARISTAS SOBRE EL VENTRÍCULO IZQUIERDO NO COMPACTO
Muchas cuestiones quedan por definir en cuanto al VINC, Arbustini las refiere en su artículo del 201421: si se trata de una MCP distinta o de un carácter morfológico compartido por distintas MCP. Se han discutido diversas estrategias para el diagnóstico y el tratamiento de estos pacientes en cuanto a embriología, mecanismos básicos, epidemiología, anatomía, patología, manifestaciones clínicas, imágenes, modalidades terapéuticas y genética34,35. Los tres marcadores que definen la enfermedad son las trabéculas prominentes del VI, los recesos intertrabeculares profundos y la fina capa compacta. Aunque existen datos genéticos en ratones y seres humanos que apoyan que se trata de una MCP distinta, hay evidencias de que es un carácter morfológico compartido con otras enfermedades. La interpretación más completa de las imágenes y los avances genéticos, llevan al mayor entendimiento de sus mecanismos básicos y de su tratamiento óptimo. La comprensión de que existe un amplio espectro de variabilidad morfológica, desde el corazón con una capa compacta casi ausente y un componente exclusivo trabecular en el ápex del VI, hasta los corazones con trabéculas prominentes y recesos profundos alternantes pero con una capa compacta bien representada. El VINC puede presentarse aislado o asociado a MCP, enfermedades congénitas y síndromes complejos que afectan al corazón. La American Heart Association clasifica el VINC como una MCP genética, mientras la European Society of Cardiology lo incluye como MCP no clasificada, al igual que la clasificación internacional de enfermedades de la Organización Mundial de la Salud.
El VINC puede ser familiar (hereditario) o no (esporádico, si se prueba que está ausente en los familiares), adquirido como en atletas de alto rendimiento, sicklémicos y embarazadas (a veces el fenotipo trabecular puede ocurrir por una carga mecánica y desaparecer cuando aquella cesa o en el posparto); se ignora si en estos casos hay un componente genético de la enfermedad. El 75% de los niños con anormalidades electrocardiográficas y muerte, tienen función sistólica deprimida; algunos de ellos con recuperación transitoria seguida de deterioro, lo cual sugiere una naturaleza genética. Muchos casos familiares se asocian a mutaciones en los mismos genes que originan otros tipos de MCP y no está claro si estos causan la MCP o si se trata de un fenotipo determinado. Aunque no existe un gold estándar para el diagnóstico del VINC, las imágenes son la mejor herramienta y se relacionan con los hallazgos en la anatomía patológica (autopsia o trasplante): el ecocardiograma es básico y la resonancia magnética nuclear ofrece detalles anatómicos y funcionales. El manejo clínico de los pacientes con VINC depende del fenotipo funcional y de las complicaciones; en el caso de las arritmias, las opciones son: los dispositivos, la ablación de focos arritmogénicos, la resincronización y la remodelación quirúrgica del VI.
Dadas las múltiples bases etiológicas del VINC, puede vérsele como un carácter aislado o una enfermedad asociada a enfermedades genéticas y defectos congénitos; esporádica o adquirida en condiciones fisiológicas o patológicas; permanente o transitoria; u originarse durante el desarrollo embriológico (hipótesis embriogénica). El proceso de trabeculación del corazón empieza después del estadio de asa del corazón, la formación de las trabéculas se inicia con la emergencia de los miocitos a través de la delaminación (migración) del miocardio compacto. La dilatación y la hipertrofia del VI pueden estar presentes o no. El VINC por sí mismo no describe necesariamente una enfermedad, puede ser una variante anatómica de la estructura del VI y su diagnóstico diferencial comprende la hipertrabeculación prominente con capa compacta normal del VI, la MCP-H apical, la MCP-D, la fibroelastosis endocárdica y los trombos apicales del VI. Si las dimensiones y la función del VI son normales, sólo se requiere un seguimiento clínico; si hay sintomatología por dilatación, disfunción o hipertrofia, el tratamiento depende de la IC, de las arritmias y del fenotipo (las pruebas genéticas no lo modifican).
Pueden presentarse IC, arritmias (fibrilación auricular, taquicardia ventricular en el 47% de los sintomáticos), episodios de MS cardíaca (13-18%), eventos embólicos sistémicos y otros. Quedan cosas por definir en el futuro: si es una enfermedad primaria, si existe aislado o asociado a otra MCP; si resulta clínicamente útil indicar el fenotipo de la MCP y el VINC (MCP-H, MCP restrictiva, MCP-D, MCP arritmogénica del ventrículo derecho), para distinguirlo del VINC aislado con diámetro y función normales del VI; su papel como marcador clínico y la hipótesis genética diagnóstica. En general se requieren criterios diagnósticos unificados reproducibles (basados en imágenes y en registros mundiales), y datos sobre la evolución de estos pacientes, con una mayor colección de casos y registros de imágenes y de información genética21.
En cuanto al tratamiento, debe llegarse al diagnóstico correcto del fenotipo por las distintas evoluciones y las diversas conductas a emplear. En el VINC hereditario puede practicarse cribado (screening) en los parientes de primer grado _que en general no se han realizado imágenes_, y las pruebas genéticas pueden afectar la conducta a seguir, que varía según exista disfunción miocárdica, arritmias, ambas, o enfermedad cardíaca congénita, con opciones muy variadas: anticongestivos, inhibidores de la enzima convertidora de angiotensina, betabloqueadores, antagonistas de la aldosterona, diuréticos, vasodilatadores, aspirina, inotropos, CDAI, resincronizador, trasplante, bloqueadores de calcio, antiagregantes o anticoagulantes, otros tratamientos con vitaminas, coenzimas, carnitina, procedimientos por cateterismo percutáneo o cirugía36.
Existen varios subtipos de VINC, al menos con 8 fenotipos distintos, con tratamientos y evoluciones diversos36 (Tabla 2).

Como se mencionó anteriormente, el electrocardiograma en el VINC usualmente es anormal. El 87% cursa con hipertrofia (del VI o biventricular) por criterios de voltaje, inversión de la onda T, anormalidades del segmento ST o sobrecarga, crecimiento auricular izquierdo, desviación axial izquierda, QT prolongado o preexcitación. En los neonatos y niños pequeños el voltaje de QRS puede ser extremo.
Las arritmias son supraventriculares y ventriculares, puede haber bradiarritmias, muchas de ellas con peligro para la vida. El subtipo con anormalidades tempranas del ritmo cursa con peligro de MS. El CDAI es muy efectivo para prevenir la MS arrítmica, con inclusión de aquellos con grave disfunción del VI, historia previa de taquicardia supraventricular o fibrilación, síncope recurrente de origen desconocido o historia familiar de MS cardíaca. Las taquiarritmias ventriculares (incluyendo aquellas con fibrilación ventricular que causan paro cardíaco), se informan en el 38-47% de los adultos con VINC y en el 13-18% de quienes mueren súbitamente36.
Una serie de 77 adultos, informa que en 44 de ellos se colocó un CDAI (por las guías estándar para las MCP no isquémicas), con un seguimiento promedio de 33 meses, 8 tuvieron choques apropiados después de 6 meses (mediana), lo que sugiere que el VINC tiene alto riesgo de MS cardíaca. Las descargas apropiadas se asocian a taquicardias ventriculares aunque a veces no se conoce el ritmo inicial en pacientes con MS cardíaca (fibrilación ventricular desencadenada por una taquicardia ventricular)36.
En los pacientes con VINC y arritmias ventriculares sostenidas, las recurrencias con choques apropiados fueron de 33% en un seguimiento medio de 26 meses. También se han informado choques apropiados en el 37% de los pacientes con VINC y CDAI en un seguimiento de 40 meses. En niños pequeños, pueden indicarse fármacos antiarrítmicos antes del CDAI por la alta frecuencia de fracturas de los electrodos y choques inapropiados en esa población36.
Los adultos pueden tener alto riesgo de taquiarritmias ventriculares y episodios de MS cardíaca, 47-74% de los sintomáticos mueren dentro de los 6 años a partir de la presentación. Investigaciones más recientes hablan de una historia natural más benigna, con menor riesgo de arritmias ventriculares. En un estudio de 241 pacientes adultos con VINC aislado, hubo un 6,2% de muerte cardiovascular con medidas asociadas (trasplante, CDAI) y un 8,6% de eventos cardiovasculares (muerte, ictus, choques del CDAI, trasplante)36.
Las arritmias ventriculares en el VINC se han relacionado con: microreentradas en el miocardio trabeculado, hipoperfusión coronaria epicárdica y déficit en el desarrollo del sistema de conducción. Se ha sugerido que las extrasístoles ventriculares en esta enfermedad se originan principalmente en el sistema de conducción y el miocardio relacionado, y no en las áreas ecocardiográficas afectadas por la no compactación. Van Malderen et al.37 estudiaron 101 pacientes con VINC para precisar el origen de las extrasístoles, compararon los segmentos afectados por la no compactación con dicho origen y encontraron que el 95% de ellas no nacía de las áreas de VINC, y el 10% tuvo un verdadero origen miocárdico. El resto se originaba en otras estructuras (tracto de salida, fascículos y anillos mitral y tricuspídeo). Identificar el mecanismo electrofisiológico básico de la arritmogénesis resulta de interés a la hora de seleccionar la terapia en estos pacientes (fármacos antiarrítmicos, EEPC, ablación)37.
Se ha informado una alta prevalencia de repolarización precoz en pacientes con VINC, en especial en aquellos con arritmias ventriculares malignas (75%) frente al 31% en los casos sin ellas. Se sabe que este hallazgo se asocia a las arritmias, incluida la fibrilación ventricular y los eventos de MS. Un posible mecanismo es la trabeculación aumentada en las invaginaciones intramiocárdicas profundas y en el sistema Purkinje profundo del miocardio medio, lo cual resulta en despolarización retardada, repolarización no homogénea y heterogeneidad transmural. Se ignora si hay factores genéticos a nivel de los canales que influyen en la vulnerabilidad a las arritmias ventriculares y a la repolarización precoz, o si se trata de un dominio de la corriente Ito del epicardio ventricular.
En el VINC está ausente la torcedura normal del VI, posiblemente por inmadurez del sistema espiral del endocardio. La taquicardia y la fibrilación ventriculares son frecuentes, Caliskan et al.38 estudiaron 84 pacientes con esta enfermedad e informaron que un 39% tuvo repolarización precoz (con localizaciones inferior en el 6%, lateral en el 27% y ambas en el 15%; no se observaron en las derivaciones V1-V3). En los casos con taquicardia-fibrilación ventriculares hubo repolarización precoz en el 75% vs. 31% en los otros pacientes. La evolución fue peor en los pacientes con estas arritmias y repolarización precoz (ésta también se observa en la fibrilación ventricular idiopática y en el síndrome de QT corto) y es más trascendente si el segmento ST es horizontal o descendente, y de menor riesgo si es ascendente rápido. Una causa de esta repolarización puede ser la mayor trabeculación del VI en las invaginaciones endomiocárdicas profundas y parece existir una posible asociación entre ambas cosas. Puede emplearse la quinidina, que restaura la homogeneidad eléctrica transmural, aborta la activación arrítmica, disminuye el patrón de repolarización precoz y disminuye o elimina las arritmias. En resumen, la repolarización precoz contribuye a estratificar riesgo en estos pacientes y a veces, ayuda a identificar a quienes necesitan CDAI38.
Existen pocos datos sobre los dispositivos CDAI en los pacientes de VINC, es una opción como profilaxis primaria o secundaria, con terapias que suelen ser apropiadas en ambos grupos. Caliskan et al.39, en otra investigación, estudiaron 77 pacientes adultos, con CDAI en 44 de ellos, según las indicaciones de las guías para las MCP no isquémicas (taquicardia y fibrilación ventriculares e IC grave). Las terapias fueron inapropiadas en la profilaxis primaria en el 19% y en la secundaria en el 25%; resultaron apropiadas en el 13% en la primaria y en el 33% en la secundaria. En los pacientes con VINC se informa la ocurrencia de taquicardia ventricular en el 38-47% y de MS en el 13-18% (incluidas la taquicardia y la fibrilación ventriculares). Según el examen histológico del miocardio que rodea los recesos intratrabeculares profundos, allí pueden crearse zonas de conducción lenta y posibles reentradas; y la mala reserva del flujo (isquemia intermitente) puede contribuir a esta arritmogenia. Estos autores consideran de poco valor la inducibilidad de una taquicardia ventricular sostenida en la EEPC para estratificar riesgo en estos pacientes y plantean que las extrasístoles no parecen asociarse a un peor pronóstico aunque los datos sobre este asunto aun son limitados39.
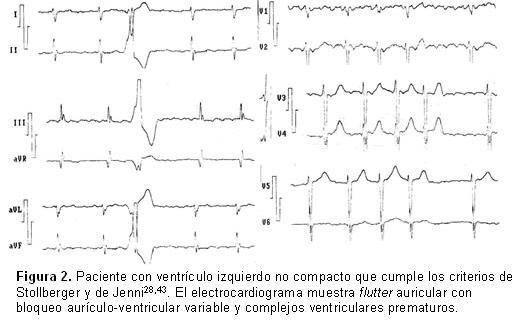
Existen pocos informes sobre los posibles beneficios de la ablación en pacientes con VINC y arritmias ventriculares. Muser et al.40 estudiaron 9 pacientes, 3 con taquicardia ventricular y 6 con extrasístoles (en estas, zonas medioapicales no compactas frente a arritmias que nacían en regiones basales septales del VI o en los músculos papilares, o en ambos), el proceso no compacto se extendió a los segmentos de los músculos papilares; en las zonas remotas al miocardio no compacto, el músculo patológico exhibía fibrosis, disrupción de la arquitectura celular y miocitos no compactos. Esta es la primera serie de ablación en arritmias ventriculares refractarias a tratamiento en el VINC y se informan buenos resultados (89% de los casos y mejoría de la fracción de eyección en el 50%) y pocas complicaciones. El acoplamiento anormal célula a célula en el miocardio no compacto, la disfunción regional microvascular y la actividad anormal de los canales iónicos resultan ideales para las reentradas y los mecanismos focales. El origen de las arritmias ventriculares se relaciona con el ventrículo no compacto sobre todo en la taquicardia ventricular, la distribución del sustrato es inusual e interesa el tracto ventricular de salida, el ápex del VI y la zona medioapical. En las figuras (Figura 2, Figura 3 y Figura 4) se presentan varios ejemplos de VINC con distintas alteraciones electrocardiográficas y arritmias.


CONFLICTOS DE INTERESES
Los autores declaran que no existen conflictos de intereses.
BIBLIOGRAFÍA
1. Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett DG, et al. Contemporary definitions and classification of the cardiomyopathies: An American Heart Association scientific statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation. 2006;113(14):1807-16.
2. Bayés de Luna A, López-Sandón J, Attie F, Alegría Ezquerra E. Cardiología clínica. Barcelona: Masson SA; 2003. p. 12.
3. Elliott PM. Classification of cardiomyopathies: Evolution or revolution. J Am Coll Cardiol. 2013;62(22):2073-4.
4. Richardson P, MacKenna W, Bristow M, Maisch B, Mautner B, O'Connell J, et al. Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the definition and classification of cardiomyopathies. Circulation. 1996;93(5):841-2.
5. Thiene G, Corrado D, Basso C. Cardiomyopathies: Is it time for a molecular classification? Eur Heart J. 2004;25(20):1772-5.
6. Ackerman MJ, Priori SG, Willems S, Berul C, Brugada R, Calkins H, et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies: This document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Europace, 2011;13(8):1077-109.
7. Chopra N, Knollmann BC. Genetics of sudden cardiac death syndromes. Curr Opin Cardiol. 2011;26(3):196-203.
8. Bayés de Luna A. Clinical arrhythmology. Oxford: Wiley-Blackwell; 2011.
9. Celiker A, Ozkutlu S, Dilber E, Karagöz T. Rhythm abnormalities in children with isolated ventricular noncompaction. Pacing Clin Electrophysiol. 2005;28(11):1198-202.
10. Arbustini E, Narula N, Dec GW, Reddy KS, Greenberg B, Kushwaha S, et al. The MOGE(S) classification for a phenotype-genotype nomenclature of cardiomyopathy: Endorsed by the World Heart Federation. J Am Coll Cardiol. 2013;62(22):2046-72.
11. Bayés de Luna A. Clinical electrocardiography. A textbook. 4ª ed. Oxford: Wiley-Blackwell; 2012. p. 453-4, 458-60.
12. Yin L. Non-compact cardiomyopathy or ventricular non-compact syndrome? J Cardiovasc Ultrasound. 2014;22(4):165-72.
13. Brescia ST, Rossano JW, Pignatelli R, Jefferies JL, Price JF, Decker JA, et al. Mortality and sudden death in pediatric left ventricular noncompaction in a tertiary referral center. Circulation. 2013;127(22):2202-8.
14. Murphy RT, Thaman R, Blanes JG, Ward D, Sevdalis E, Papra E, et al. Natural history and familial characteristics of isolated left ventricular non-compaction. Eur Heart J. 2005;26(2):187-92.
15. Kobza R, Jenni R, Erne P, Oechslin E, Duru F. Implantable cardioverter-defibrillators in patients with left ventricular noncompaction. Pacing Clin Electrophysiol. 2008;31(4):461-7.
16. Ikeda U, Minamisawa M, Koyama J. Isolated left ventricular non-compaction cardiomyopathy in adults. J Cardiol. 2015;65(2):91-7.
17. Thiene G, Corrado D, Basso C. Revisiting definition and classification of cardiomyopathies in the era of molecular medicine. Eur Heart J. 2008;29(2):144-6.
18. Arbustini E, Narula N, Dec GW, Reddy KS, Greenberg B, Kushwaha S, et al. Reply: The MOGE(S) classification for a phenotype-genotype nomenclature of cardiomyopathy: More questions than answers? J Am Coll Cardiol. 2014;63(23):2584-6.
19. Pasotti M. The MOGE(S) classification for a phenotype-genotype nomenclature of cardiomyopathy: More questions than answers? J Am Coll Cardiol. 2014;63(23):2584.
20. Arbustini E, Narula N, Tavazzi L, Serio A, Grasso M, Favalli V, et al. The MOGE(S) classification of cardiomyopathy for clinicians. J Am Coll Cardiol. 2014;64(3):304-18.
21. Arbustini E, Weidemann F, Hall JL. Left ventricular noncompaction: A distinct cardiomyopathy or a trait shared by different cardiac diseases? J Am Coll Cardiol. 2014;64(17):1840-50.
22. Steffel J, Kobza R, Oechslin E, Jenni R, Duru F. Electrocardiographic characteristics at initial diagnosis in patients with isolated left ventricular noncompaction. Am J Cardiol. 2009;104(7):984-9.
23. Sen-Chowdhry S, McKenna WJ. Left ventricular noncompaction and cardiomyopathy: Cause, contributor, or epiphenomenon. Curr Opin Cardiol. 2008;23(3):171-5.
24. Duru F, Candinas R. Noncompaction of ventricular myocardium and arrhythmias. J Cardiovasc Electrophysiol. 2000;11(4):493.
25. Modi S, Krahn AD. Sudden cardiac arrest without overt heart disease. Circulation. 2011;123(25):2994-3008.
26. Stollberger C, Gerger D, Jirak P, Wegner C, Finsterer J. Evolution of electrocardiographic abnormalities in association with neuromuscular disorders and survival in left ventricular hypertrabeculation/noncompaction. Ann Noninvasive Electrocardiol. 2014;19(6):567-73.
27. Viskin S, Rogowski O. Asymptomatic Brugada syndrome: A cardiac ticking time-bomb? Europace. 2007;9(9):707-10.
28. Dorantes M, Pham Trung C. Estimulación eléctrica programada del corazón en el síndrome de Brugada. Parte II: Variaciones sobre un mismo tema. CorSalud [Internet]. 2015 [citado 30 Sep 2017];7(3):202-13. Disponible en: http://www.revcorsalud.sld.cu/index.php/cors/article/view/59/96
29. Dorantes M, Pham Trung C. Estimulación eléctrica programada del corazón en el síndrome de Brugada. Parte I: Una visión actual. CorSalud [Internet]. 2015 [citado 30 Sep 2017];7(1):46-51. Disponible en: http://www.revcorsalud.sld.cu/index.php/cors/article/view/11/11
30. Josephson ME. Electrophysiology at a crossroads. Heart Rhythm. 2007;4(5):658-61.
31. Josephson ME. Electrophysiology at a crossroads: A revisit. Heart Rhythm. 2016;13(12):2317-22.
32. Buxton AE. Programmed ventricular stimulation: not dead. Circulation. 2014;129(8):831-3.
33. Martini B, Martini N, Dorantes Sánchez M, Márquez MF, Zhang L, Fontaine G, et al. Pistas de una enfermedad orgánica subyacente en el síndrome de Brugada. Arch Cardiol Mex. 2017;87(1):49-60.
34. Tester DJ, Ackerman MJ. Genetic testing for potentially lethal, highly treatable inherited cardiomyopathies/channelopathies in clinical practice. Circulation. 2011;123(9):1021-37.
35. Celiker A, Kafali G, Doðan R. Cardioverter defibrillator implantation in a child with isolated noncompaction of the ventricular myocardium and ventricular fibrillation. Pacing Clin Electrophysiol. 2004;27(1):104-8.
36. Towbin JA, Lorts A, Jefferies JL. Left ventricular non-compaction cardiomiopathy. Lancet. 2015;386(9995):813-25.
37. Van Malderen S, Wijchers S, Akca F, Caliskan K, Szili-Torok T. Mismatch between the origin of premature ventricular complexes and the noncompacted myocardium in patients with noncompaction cardiomyopathy patients: involvement of the conduction system? Ann Noninvasive Electrocardiol [Internet]. 2017 [citado 30 Sep 2017];22(2):e12394. Disponible en: https://doi.org/10.1111/anec.12394
38. Caliskan K, Ujvari B, Bauernfeind T, Theuns DA, Van Domburg RT, Akca F, et al. The prevalence of early repolarization in patients with noncompaction cardiomyopathy presenting with malignant ventricular arrhythmias. J Cardiovasc Electrophysiol. 2012;23(9):938-44.
39. Caliskan K, Szili-Torok T, Theuns DA, Kardos A, Geleijnse ML, Balk AH, et al. Indications and outcome of implantable cardioverter-defibrillators for primary and secondary prophylaxis in patients with noncompaction cardiomyopathy. J Cardiovasc Electrophysiol. 2011;22(8):898-904.
40. Muser D, Liang JJ, Witschey WR, Pathak RK, Castro S, Magnani S, et al. Ventricular arrhythmias associated with left ventricular noncompaction: Electrophysiologic characteristics, mapping, and ablation. Heart Rhythm. 2017;14(2):166-75.
41. Viskin S. The QT interval: too long, too short or just right. Heart Rhythm. 2009;6(5):711-5.
42. Anttonen O, Junttila MJ, Maury P, Schimpf R, Wolpert C, Borggrefe M, et al. Differences in twelve-lead electrocardiogram between symptomatic and asymptomatic subjects with short QT interval. Heart Rhythm. 2009;6(2):267-71.
43. Jerez AM, Echevarría S, Guevara G, Aleaga E. Miocardiopatía por ventrículo no compactado. Acerca de su evolución histórica, definiciones y generalidades del tema. Rev Cubana Cardiol Cir Cardiovasc [Internet]. 2015 [citado 30 Sep 2017];21(2):117-22. Disponible en: http://www.revcardiologia.sld.cu/index.php /revcardiologia/article/view/586/pdf_11
Recibido: 03 de octubre de 2017
Aceptado: 23 de noviembre de 2017
Margarita Dorantes Sánchez. Servicio de Arritmias y Estimulación Eléctrica Cardíaca. Instituto de Cardiología y Cirugía Cardiovascular. La Habana, Cuba. Correo electrónico: dorantes@infomed.sld.cu

