










 Servicios personalizados
Servicios personalizados Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
Desde que la displasia arritmogénica del ventrículo derecho (DAVD) fue descrita por primera vez por Dalla Volta et al1 en 1961, y posteriormente caracterizada por Fontaine et al2 en 1977, hasta la actualidad, en la que ha sido incluida en la clasificación de la Organización Mundial de la Salud de las miocardiopatías3, las aportaciones a la literatura acerca de esta enfermedad han sido numerosas4,5. Inicialmente las descripciones se centraban en el sustrato arrítmico de ciertas zonas del ventrículo derecho, el llamado «triángulo de la displasia»; pero actualmente el espectro se ha ampliado para dar paso a manifestaciones difusas en el mencionado ventrículo, a la afección única ventricular izquierda y biventricular en fase avanzada de la enfermedad, a menudo indistinguible de la miocardiopatía dilatada6. La prevalencia varía ampliamente según las series descritas y existe controversia acerca de la distribución geográfica de la enfermedad. En regiones italianas del Véneto se estima una prevalencia de 1 caso por 1000 o 10000 personas. Corrado et al7 plantean que puede ser causa de hasta el 20% de las muertes súbitas cardíacas (MSC) en adultos jóvenes y en atletas italianos, y afecta más frecuentemente a los varones. En EEUU representa un 5% de las MSC en menores de 65 años y 3-4%, en atletas8,9.
La DAVD es una enfermedad del músculo cardíaco de origen genético cuyo diagnóstico supone a menudo un reto para el clínico. La descripción clásica suele referirse al estadio final de la enfermedad, en que el miocardio, fundamentalmente del ventrículo derecho, ha sido sustituido por tejido fibroadiposo; por eso las fases iniciales de la enfermedad, no tan floridas en semiología, suelen pasar inadvertidas. Desafortunadamente, el riesgo de desenlace fatal no es bajo10.
CASO CLÍNICO
Se trata de un hombre de piel blanca y 54 años de edad, sin factores de riesgo cardiovascular, hábitos tóxicos, enfermedad valvular previa u otra cardiopatía, que fue remitido a consulta de arritmia por presentar episodios de pérdida de la conciencia, de breve duración, relacionadas con el esfuerzo físico y situaciones de estrés, y finalmente taquicardia ventricular (TV), lo que degeneró en fibrilación ventricular recuperada tras cardioversión eléctrica. El ECG basal de doce derivaciones presentaba patrón de bloqueo de rama derecha incompleto con ondas T negativas y anchura del QRS mayor de 110 mseg; además, onda épsilon y fragmentación del QRS en precordiales derechas (Figura 1). El ecocardiograma realizado demostró dilatación del ventrículo derecho (54 mm) con pared libre engrosada y presencia de zonas de aspecto blanquecino, que pudieran estar en relación con infiltración fibroadiposa (Figura 2). Se diagnosticó una DAVD, se indicó tratamiento con amiodarona y se le colocó un desfibrilador automático implantable, con lo que evolucionó favorablemente.
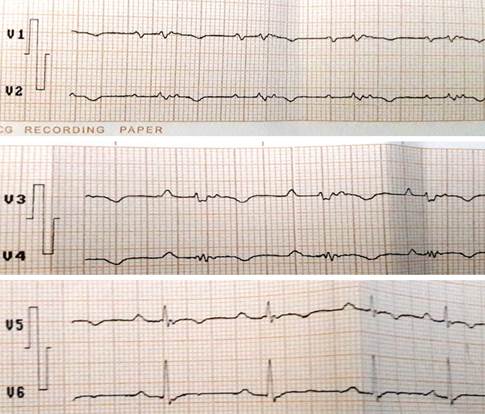
Figura 1 Electrocardiograma en precordiales derechas con onda épsilon y QRS ancho, con fragmentación.

COMENTARIOS
La DAVD es un trastorno heredable, que tiene clara incidencia familiar hasta en el 50% de los casos, con un patrón de transmisión autosómico dominante, diversos grados de penetración y una expresión fenotípica polimórfica. No obstante, también se ha descrito una forma autosómica recesiva1-5. Se puede asociar a una queratodermia palmo-plantar y a un pelo lanudo (enfermedad de Naxos)6. Este tipo de DAVD lo causa una mutación en el gen de la placoglobina, cuyo producto es un componente de los desmosomas y las uniones celulares adherentes (adherens), y el síndrome de Carvajal, con el mismo fenotipo cutáneo a predominio del ventrículo izquierdo en familias de la India y Ecuador6,7.
La primera mutación causante de una DAVD no sindrómica fue descrita por Rampazzo et al8 en 2002. Dicha mutación se identificó en el gen de la desmoplaquina, que codifica un componente del desmosoma. En 2004, Gerull et al9 describieron 25 mutaciones del gen desmosómico cardíaco de la placofilina; posteriormente, se identificaron otras mutaciones presuntamente causantes de enfermedad en la placoglobina y la desmoplaquina, así como en otros genes desmosómicos (los de la desmocolina 2 y la desmogleína 2) en pacientes con DAVD no sindrómica. Actualmente se considera que la disfunción desmosómica es la vía final común en la patogenia de la DAVD, ya que la integridad estructural y funcional del tejido cardíaco se basa en los desmosomas, las uniones adherentes (tipo adherens) y las uniones estrechas (tipo gap) situadas en los discos intercalares10.
En el mapa cromosómico se han localizado diferentes variantes genéticas de DAVD y se han descrito más de 140 mutaciones causantes de enfermedad, la mayoría de ellas correspondientes a genes que codifican proteínas desmosómicas. Algunos genes no desmosómicos se han asociado también a una DAVD autosómica dominante, entre ellos el gen del factor de crecimiento transformador β-3 (TGFβ3)11, que modula la expresión de las proteínas de contacto celular y el gen del receptor de rianodina 2 (RyR2)11,12. El gen RyR2, que se describió primero en ocho familias, codifica receptores que intervienen en la liberación del calcio del retículo sarcoplásmico; sin embargo, sigue habiendo opiniones divergentes en cuanto a si debe considerarse o no que los pacientes con mutaciones de este gen sufren una DAVD o una TV polimórfica catecolaminérgica. La mutación que se ha descrito de forma más reciente afecta al gen TMEM43 y causa una variante de DAVD de penetración completa, muy letal (DAVD tipo 5)13-15.
La degeneración y la muerte de los miocardiocitos son la consecuencia anatomopatológica de estas mutaciones de proteínas de adhesión, con la consiguiente sustitución progresiva por tejido adiposo y fibroadiposo. En la teoría inflamatoria, apoyada por la aparición de infiltrados inflamatorios en series necrópsicas, el daño miocárdico vendría explicado por un proceso continuado de daño y reparación que una miocarditis crónica13-15.
Las manifestaciones clínicas de la DAVD son variables y dependen de la inestabilidad cardíaca y la disfunción ventricular progresiva. Varían desde pacientes asintomáticos, MSC como primera manifestación, arritmias ventriculares y supraventriculares, hasta insuficiencia cardíaca derecha o biventricular. Se ha descrito la presencia de un desequilibrio en la inervación adrenérgica como posible coadyuvante en la génesis de las arritmias; de este modo, la propensión a arritmias ventriculares aumenta en situaciones de exposición a las catecolaminas, especialmente durante el ejercicio11.
Las alteraciones más frecuentes en el electrocardiograma (ECG) son la inversión de la onda T (V1-V3), presente hasta en el 50% de los sujetos. La afección más allá de V3 indica afección adicional del ventrículo izquierdo12. Existen diferentes anomalías de la despolarización ventricular, el bloqueo de rama derecha incompleto es más frecuente (18%) que el completo (15%); la prolongación del QRS más de 110 milisegundos en V1 y V2 es un hallazgo más específico, y pueden aparecer ondas épsilon que se observan al final del QRS y al inicio del ST, y corresponden a potenciales eléctricos retrasados de pequeña amplitud originados en las áreas de tejido sano rodeadas de infiltrado fibroadiposo13. La anchura del QRS y su fragmentación en las precordiales derechas permiten predecir, de manera independiente, la presencia de dilatación y disfunción del ventrículo derecho e incluso la aparición de arritmias14.
Esta fragmentación del QRS se define como la presencia de muescas u ondas de bajo voltaje (R’) en la porción terminal del QRS o en el inicio del segmento ST, en al menos dos derivaciones contiguas. Morita et al10 demostraron que constituyen un marcador de la presencia de un sustrato propicio para el surgimiento de fibrilación ventricular espontánea, con una sensibilidad de 93% y especificidad de 90%.
El diagnóstico definitivo de la DAVD requiere la confirmación anatomopatológica de la sustitución fibroadiposa transmural, mediante muestras quirúrgicas o necrópsicas. La naturaleza parcheada y progresiva de la enfermedad hace que la biopsia endomiocárdica tenga una utilidad limitada. No existe una única prueba para establecer el diagnóstico de DAVD3-7; éste se establece tras una evaluación funcional, morfológica y electrocardiográfica, mediante la cual se determinan los criterios mayores y menores actualmente reconocidos15-17. En 2002 se propuso una modificación de estos criterios para el diagnóstico de la DAVD, en familiares de primer grado de un caso inicial16. En esta situación, la presencia de una inversión de la onda T en precordiales derechas (V2-V3), potenciales tardíos en el ECG de promediación de señal, TV con morfología de bloqueo de rama izquierda del haz de His (BRIHH), o la observación de cambios funcionales o morfológicos del ventrículo derecho en las exploraciones de imagen, deben considerarse criterios mayores con valor diagnóstico para la DAVD familiar16. Posteriormente se publicó una nueva modificación de estos criterios con la finalidad de aumentar la sensibilidad mediante el uso de las modalidades diagnósticas emergentes, los avances en la genética de la DAVD y la introducción de parámetros cuantitativos18.
La DAVD se manifiesta generalmente en forma de episodios de TV originada en el VD, por lo que tienen morfología de BRIHH, en adolescentes o adultos jóvenes aparentemente sanos. Las arritmias ventriculares pueden ser asintomáticas y detectarse en un ECG sistemático o pueden causar palpitaciones, síncope o MSC. Se ha estimado que la DAVD explica hasta un 5-20% de los casos de MSC en individuos de menos de 35 años de edad18.
Aunque la información en relación con la historia natural es limitada, en general se admiten 4 estadios7,19,20:
La fase temprana o silente, generalmente asintomática, aunque el debut puede manifestarse con MSC.
Fase inestable con predominio de arritmias sintomáticas, generalmente con morfología de BRIHH, altamente sugestivas de origen ventricular derecho.
Fase de fallo ventricular derecho con relativa conservación de la función del izquierdo.
Fase final con progresiva dilatación biventricular, a menudo indistinguible de la miocardiopatía dilatada. Las complicaciones más frecuentes en este estadio son las tromboembólicas y la fibrilación auricular.
En las últimas dos décadas, las arritmias originadas en el VD, han atraído la atención del mundo científico por diversas razones, principalmente porque suelen afectar a pacientes de menor edad y pueden conducir a la MSC. El mecanismo fisiopatológico de esas arritmias no se ha aclarado por completo y a veces deja margen para diferentes interpretaciones. Además, cada vez se involucra en mayor medida al intrigante mundo de la genética en los aspectos patogénicos, diagnósticos y pronósticos de algunas de estas arritmias. La infiltración por tejido fibroadiposo constituye un sustrato para la inestabilidad eléctrica y lleva a la arritmia ventricular que va desde las extrasístoles ventriculares aisladas hasta las TV sostenidas o la fibrilación ventricular1-4. Existen múltiples predictores eléctricos para la aparición de las arritmias ventriculares y MCS10,21-23, el QRS de mayor duración, la fragmentación del QRS y la onda épsilon estuvieron presentes en nuestro paciente.
El objetivo principal de la estrategia terapéutica en este tipo de pacientes es la prevención de la MSC, para lo cual disponemos de 3 estrategias terapéuticas principales: los fármacos antiarrítmicos, la ablación por catéter y el uso del desfibrilador automático implantable24-26.

