










 Servicios personalizados
Servicios personalizados Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
Se define como defecto congénito a toda alteración de estructura anatómica visible al examen clínico del recién nacido, o posterior al nacimiento, cuando se percibe el defecto funcional de un órgano interno afectado anatómicamente1.
Las cardiopatías congénitas son todos los defectos congénitos cardíacos que se producen como consecuencia de alteraciones morfológicas durante el proceso de organogénesis. La mayoría son bien toleradas durante la vida intrauterina y se manifiestan después del nacimiento, cuando se pierde el contacto con la circulación materna. Existen diferentes clasificaciones, basadas en criterios clínicos (cianóticas y acianóticas), genéticos (sindrómicas y no sindrómicas), pronóstico (críticas, potencialmente críticas y no críticas), y según la presencia o no de defectos congénitos extracardíacos (aisladas y asociadas). El Registro Cubano de Malformaciones Congénitas (RECUMAC) las clasifica en simples y complejas, mientras que la propuesta por Hoffman y Kaplan se basa en la gravedad del defecto congénito1-3.
Las cardiopatías congénitas constituyen los defectos congénitos más frecuentes, y afectan aproximadamente entre 6 a 11 neonatos por cada 1000 nacidos vivos. Entre 20% y 30% de estas cardiopatías son graves y se ha informado su incremento en estudios recientes, específicamente defectos septales sin que existan variaciones relevantes en las prevalencias de las más graves, lo que está relacionado con un mayor nivel diagnóstico4.
Se desconocen las causas en un gran número de casos, aunque existen evidencias de que los factores genéticos desempeñan un papel decisivo en aproximadamente un 8% de los casos y los agentes ambientales están involucrados entre 1-2% de ellos, la génesis del resto (90%) es multifactorial y resulta de una compleja interrelación entre la predisposición genética, la susceptibilidad epigenética, el ambiente parental y el estilo de vida.
Entre los síndromes genéticos que cursan con cardiopatías congénitas se describen los monogénicos (3-5%), cromosómicos (5-8%), microdeleciones y algunos por mutaciones en la molécula de ácido desoxirribonucleico (ADN) mitocondrial. Los desbalances génicos detectados por cariotipo convencional o por estudios de citogenética molecular explican entre el 9% y el 13% de todos los casos de cardiopatías congénitas en el período neonatal5-7.
El desarrollo de las tecnologías de secuenciación de nueva generación (NGS, del inglés Next Generation Sequencing) ha emergido como una poderosa herramienta para estudiar la secuencia de todo el exoma humano mediante la técnica denominada WES (del inglés Whole-exome sequencing) o la secuenciación de exones específicos en enfermedades monogénicas de interés. Estas dos metodologías de genética molecular han revolucionado la comprensión de las bases moleculares de muchos defectos congénitos, como las cardiopatías congénitas, al proveer una información más detallada de las variaciones génicas que las que aportan los amplios estudios de asociación genómica en el estudio de polimorfismos de un simple nucleótido (SNP, del inglés Single Nucleotide Polymorphisms)8-12.
Muchos estudios indican que el desarrollo cardíaco está estrechamente regulado por una serie de vías de señalización celular que modulan la proliferación, la migración y la diferenciación celulares en el proceso del desarrollo cardiovascular. Mutaciones en diferentes genes que participan en estas vías de comunicación intercelular subyacen en las bases de diferentes tipos de cardiopatías congénitas13-17.
MÉTODO
Se realizó una revisión de la literatura médica nacional e internacional publicada en idiomas español e inglés, la información se recopiló a través de buscadores como Google académico, las bases de datos Medline/PubMed, Bireme (Scielo, Lilacs) y la biblioteca médica Cochrane en Octubre de 2018, con el objetivo de identificar los avances en el conocimiento de las bases moleculares y celulares de las cardiopatías congénitas. La revisión bibliográfica se realizó en función de criterios cronológicos y temáticos, para la cual se seleccionaron los de mayor nivel de actualización y pertinencia.
BASES MOLECULARES Y CELULARES DEL PROCESO DE MORFOGÉNESIS CARDÍACA
Las células musculares cardíacas o miocardiocitos se diferencian en etapas tempranas del desarrollo de un grupo de células progenitoras localizadas en la porción anterior de la placa mesodérmica lateral. El corazón y los grandes vasos se forman a partir de las células mesenquimatosas del área cardiogénica, la primera señal es la aparición de bandas endoteliales pares llamadas «cordones angioblásticos» en el mesodermo cardiogénico durante la tercera semana de gestación. Estos cordones se canalizan para formar conductos longitudinales emparejados, tapizados de endotelio, denominados «tubos cardíacos endocárdicos»; los cuales, conforme tiene lugar el plegamiento embrionario lateral, migran a la línea media, se aproximan entre sí y se fusionan para formar el tubo cardíaco primordial, que para entonces ya posee dos capas de tejido: una interna de tejido endotelial y una capa externa de células miocárdicas5-7.
La fusión de los tubos cardíacos comienza en el extremo cefálico del corazón en desarrollo y progresa en dirección caudal. Al final de la tercera semana el corazón comienza a latir (entre los 22 y 23 días) y la sangre a circular. Los fenómenos eléctricos y contráctiles que suceden en el miocardiocito son controlados por genes que codifican proteínas transmembranales encargadas de transportar iones a través de la membrana celular. Estos canales iónicos forman complejos macromoleculares que incluyen una unidad principal formadora del poro de los canales de sodio y de potasio, y proteínas auxiliares que lo regulan5-7.
Este corazón primitivo sufre una torsión y rotación hacia la derecha alrededor de la cuarta semana de gestación, lo cual posiciona a las aurículas por encima de los ventrículos, asimismo a partir del tracto de salida comienzan a emerger las arterias del arco aórtico.
Alrededor de la quinta y sexta semanas de desarrollo embrionario, se forman los septos cardíacos para dividir al corazón en cuatro cámaras, y el tracto de salida o conducto arterioso se divide en la arteria pulmonar y la aorta, lo que resulta en la división de la circulación en pulmonar y sistémica, respectivamente. Más tarde ocurre una reestructuración valvular intensiva junto con el crecimiento de los ventrículos para completar la maduración del corazón. El establecimiento de la simetría izquierda-derecha es muy importante para el desarrollo normal del corazón18.
El músculo de la aurícula y ventrículos primitivos es continuo. La aurícula actúa como marcapasos cardíaco transitorio, hasta que el seno venoso asume esa función. El nodo sinusal aparece durante la quinta semana de desarrollo, inicialmente en la pared del seno venoso, pero luego se incorpora a la pared de la aurícula derecha, cerca de la entrada de la vena cava superior. Junto con células de la región aurículo-ventricular (AV) forman el nodo y fascículo AV, localizado justo por encima de los cojinetes endocárdicos. Las fibras que nacen del fascículo AV pasan de la aurícula al ventrículo y se dividen en dos ramas, derecha e izquierda, que se distribuyen por todo el miocardio ventricular.
El sistema cardiovascular es el primer sistema orgánico que alcanza un estado funcional, este desarrollo cardíaco precoz es necesario debido a que el rápido crecimiento del embrión en desarrollo no puede suplir sus necesidades nutricionales y de oxígeno, sólo mediante el proceso de difusión18.
Aunque el corazón de diferentes especies de vertebrados puede tener entre dos y cuatro cámaras, las diferentes etapas del proceso de morfogénesis están altamente conservadas a nivel molecular y celular. Los morfógenos son moléculas difusibles que especifican el tipo de célula generada en una localización anatómica específica y dirigen la migración celular y sus procesos hasta su destino definitivo, entre estos se distinguen el ácido retinoico, el factor beta transformante del crecimiento, las proteínas morfogénicas óseas, la vía de comunicación intercelular Hedgehog y la familia de proteínas Wnt2,19.
El ácido retinoico es la forma bioactiva de la vitamina A, que a nivel molecular se une a sus receptores intracelulares, los activa, y regula la expresión génica «secuencia abajo». Los genes Hox son dianas cruciales de los receptores de este ácido en el proceso de morfogénesis.
La superfamilia del factor beta de trasformación del crecimiento está formada por más de treinta miembros, entre los cuales se encuentran los factores beta de trasformación del crecimiento (TGFB, del inglés Transforming Growth Factor Beta) y las proteínas morfogénicas óseas (BMP, del inglés Bone Morphogenic Proteins). La superfamilia TGFB desempeña un importante papel en el desarrollo embrionario, la diferenciación celular y la organogénesis, y la evidencia filogenética sugiere que esta es una de las más antiguas vías de señalización intercelular, al estimarse su surgimiento hace 1,3 billones de años, antes de la divergencia evolutiva de artrópodos y vertebrados. Estudios realizados en vertebrados demuestran que al menos tres miembros de la familia BMP se expresan en la cardiogénesis. BMP-2 se expresa en el endodermo lateral, mientras que BMP-4 y 7 se expresan en el ectodermo inmediato adyacente al mesodermo precardíaco. Las BMP, de conjunto con los factores de crecimiento de los fibroblastos (FGF, del inglés Fibroblastic Growth Factor), promueven la inducción cardiogénica y regulan el tamaño de los campos cardíacos, lo que confirma la importancia de esta vía de señalización intercelular en el proceso de morfogénesis2,3,13,18.
Esta señalización es el proceso mediante el cual las células intercambian mensajes químicos que modulan el funcionamiento intracelular y dan lugar a respuestas específicas tendientes a promover la adaptación de la totalidad del organismo en un medio ambiente cambiante. La unión de una molécula de señalización extracelular (ligando) con su receptor en una célula diana, desencadena una respuesta específica que consiste en una serie de eventos moleculares mutuos activadores o inhibidores.
Los sistemas de señalización intercelular son fundamentales en el control de la expresión génica y de la función de las proteínas. Estos sistemas serán los que controlen dónde, cuándo, cuánto y por cuánto tiempo se expresan las moléculas de ácido ribonucleico (RNA). En el caso de las proteínas, controlan además, los cambios de localización, el tráfico de proteínas dentro de una célula, cómo se degradan, y las interacciones funcionales que establecen. De forma coherente con este decisivo papel en la función de los organismos, se estima que más del 20% de los genes del genoma humano codifican proteínas implicadas en la transducción de señales20.
La vía de señalización intercelular Wnt es una intrincada red de señales cuyas funciones también se han mantenido a través de la evolución de las especies. La familia Wnt comprende un grupo de genes de señalización que tiene una función muy importante en la organogénesis. Este grupo de genes determina la estructura interna de un órgano o la detención del crecimiento cuando el órgano ha alcanzado su tamaño apropiado. Otras funciones en las que se involucra a estos genes incluyen la polaridad celular, la reestructuración celular y el desarrollo axial. El término Wnt deriva de la contracción de los nombres wingless (del inglés, sin alas) e Integrase-1 (Int-1). Con el primero se acuñó al gen del segmento de polaridad cuya ausencia genera un fenotipo sin alas en la mosca de la fruta, Drosophila melanogaster, y el segundo dio el nombre a una línea celular de ratón transformada con el virus Maloney que provocaba tumor mamario2,18.
Por su parte, los más de 12 genes que integran la vía de señalización Hedgehog, forman una red, más que una vía unidireccional. La señal Hedgehog (Hh, del inglés erizo), debe su nombre a que el gen que codifica para Hh fue identificado por estudios de mutagénesis en la mosca Drosophila, donde los embriones mutantes mostraban un fenotipo con puntas desorganizadas en el exoesqueleto semejantes a las púas de los erizos. En mamíferos se han identificado tres miembros de esta familia de proteínas: Sonic hedgehog (Shh), Indian hedgehog (Ihh) y Desert hedgehog (Dhh) que participan en el proceso de morfogénesis19-21.
Se ha generado un cambio drástico en el modo de estudiar el desarrollo cardíaco con las nuevas estrategias para el estudio de la cardiogénesis, lo que ha implicado el paso de los cortes secuenciales de corazones embrionarios de distintos animales, a la aplicación de los métodos moleculares de identificación de linajes celulares, al experimentar con modelos transgénicos y análisis clonal retrospectivo. Estas tecnologías han dado una visión más dinámica del desarrollo del aparato cardiovascular y han permitido encontrar el origen, a veces insospechado, de varias estructuras anatómicas a partir de determinados grupos celulares, así como los genes y sus productos proteicos9-12,22.
Estudios realizados en embriones de ratón y de pollo han permitido esclarecer las bases moleculares del proceso cardiogénico, así se ha demostrado la presencia de dos genes básicos hélice-bucle-hélice (bHLH, del inglés Basic helix-loop-helix) en los tubos endocárdicos primitivos y en etapas más avanzadas de la morfogénesis cardíaca: HAND1 y HAND2 (del inglés heart and neural crest derivatives expressed transcript 1 and 2). Los genes bHLH son una clase de factores de transcripción (FT) que regulan la determinación del destino y la diferenciación celulares en muchos tejidos diferentes durante el desarrollo embrionario3,6,14.
La formación del sistema cardiovascular está estrictamente controlada por una red regulatoria compuesta por muchas vías de señalización intercelular, FT, entre otros. Las células del primordio cardíaco comienzan a expresar genes característicos de miocardio, como NKX2-5 y GATA4. La expresión del primero de estos FT depende de proteínas presentes en el endodermo subyacente, tales como cerberus, la BMP, FGF8 y FGF129,18,20.
Existen más de 50 factores de crecimiento, conocidos también como citoquinas, que comprenden un numeroso grupo de proteínas que actúan como mensajeros, cada uno de los cuales se une con una gran especificidad a una proteína receptora de la superficie celular, lo que activa a diferentes proteínas intracelulares de transducción de señales. Actúan principalmente sobre la proliferación celular, aunque tienen también otras funciones, como la diferenciación, la supervivencia y la migración celulares. Muchos de los sistemas de transducción de señales usados por los factores de crecimiento para transferir información hacia el núcleo y modular la transcripción génica lo hacen a través de la actividad de FT.
Estos FT son productos proteicos codificados por genes de transcripción. Se calcula que la regulación de la transcripción se basa en al menos 2000 de estos factores proteicos codificados en el genoma de los mamíferos, los cuales se unen a una secuencia de nucleótidos específica en las regiones promotora/amplificadora de genes diana y regulan la velocidad del proceso por el cual un gen se transcribe a una secuencia complementaria de ácido ribonucleico (ARN), lo que inicia el proceso de producción de una proteína, por lo que desempeñan un papel fundamental en el control de la organogénesis. Además, micro-ARN específicos tienen una función fundamental en la morfogénesis cardíaca al coordinar los patrones y niveles de expresión de los FT20.
Los genes expresados en la placa cardiogénica como TBX1, TBX5, NKX2-5, GATA4, HAND2, CASP3, TFAP2, FGF12, LBX1, MYH11, CRKL, PDLIM3 y TXNRD2 codifican para FT que se expresan en etapas tempranas del desarrollo del corazón y que son cruciales para la activación de genes cardíacos específicos y constituyen el centro regulador de la red de morfogénesis cardíaca, la cual controla la rotación del tubo cardíaco, la simetría izquierda-derecha y la formación de las cámaras cardíacas. Los genes TBX5 y NKX2-5 se unen de forma sinérgica al promotor del gen que codifica para el péptido precursor natriurético cardioespecífico tipo A (Nppa, del inglés cardiac-specific natriuretic peptide precursor type A) y de esta forma activan a este gen que es índice el desarrollo cardíaco23,24.
Los genes TBX son una familia génica con un alto grado de conservación filogenética y que tienen un dominio de fijación al ADN común, denominado T-box, y codifican para FT que intervienen en la regulación del proceso de morfogénesis cardiovascular, particularmente en la elongación del tubo cardíaco en el polo anterior y el mantenimiento de las células precursoras mesenquimatosas para la formación del tracto de salida miocardializado y tabicado. La reducción de la expresión del TBX1, que se produce en el estado de deleción hemicigota al que a menudo se denomina haploinsuficiencia, influye de manera importante en la expresión génica inicial que interviene en la morfogénesis cardíaca25.
Por su parte, NKX2-5 (NK2 homeobox 5) y LBX1 pertenecen a un grupo de genes que codifican para FT con dominio homeobox (una secuencia de 180 pares de bases conservada en diferentes especies), los cuales poseen un papel importante en la regulación de la expresión genética tejido-específica, esencial para la diferenciación tisular, así como en la determinación de patrones temporales y espaciales durante el desarrollo. Estos FT resultan críticos para la formación cardíaca en Drosophila, ya que su ausencia impide la formación del asa y la diferenciación de los ventrículos19,20,23,25.
Otras proteínas que actúan como FT, son las denominadas «en dedos de cinc», ya que se estructuran alrededor de este ión, de modo que se producen prolongaciones digitiformes y, de esta forma, se mantiene fija con una hélice, que reconoce por el surco mayor una secuencia de ADN, y corresponde a promotores de genes específicos. A este grupo pertenece el FT GATA4. El modelo mutante para este gen presenta defectos en el plegamiento anterior del embrión: no hay fusión del tubo cardíaco, y se presenta la muerte, quizá debido a la importancia de GATA4 en la señalización para la migración ventral24,25.
Recientes estudios de secuenciación exómica en casos con cardiopatías congénitas han revelado genes que codifican para proteínas con dos tipos fundamentales de funciones moleculares (las de unión a la cromatina y las que forman el complejo de FT) y desempeñan un papel fundamental en la regulación de los genes críticos del desarrollo cardiovascular. En un análisis de las variaciones en el número de «copias» mediante estudio genómico amplio de 154 casos con comunicación interventricular, An et al23 identificaron mutaciones de novo en siete genes (PRKCB, HIRA, SOX2, ING2, TP63, BCL6 y PAX3) que intervienen en la vía de unión a la cromatina, y entre los FT con efecto confirmado en la cardiogénesis se incluyen LBX1, TBX1, EN1, SOX2 y PAX3. Entre los tipos de mutaciones observaron duplicaciones en los genes PAX3 y LBX1 y deleciones en CRKL, GP1BB, PDLIM3, TBX1 y TXNRD2. Además de estas dos funciones moleculares, muchos de estos genes codifican proteínas con importantes funciones biológicas, como el proceso de morfogénesis cardíaca y la regulación de la proliferación celular, entre otras (Tabla 1).
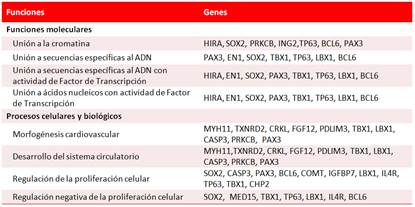
El gen PRKCB codifica para una proteína quinasa y el HIRA lo hace para una histona que interviene en la regulación del ciclo celular, mientras que el gen SOX2 pertenece a los genes que producen alta movilidad (HMG, del inglés high movility group) y contienen una secuencia, conservada evolutivamente, de 79 aminoácidos, que dan lugar a tres estructuras helicoidales, una de las cuales está inmovilizada y penetra en el surco menor del ADN, en la zona del promotor, y le provoca una flexión que facilita el acercamiento de otras proteínas al sitio de inicio de la transcripción. Mientras que la familia de genes PAX consta de al menos nueve miembros (PAX1-9) que codifican proteínas y que son FT que tienen en común el dominio de unión al ADN que se encuentra hacia el extremo amino-terminal (dominio de cajas pareadas (PAX, del inglés paired box). PAX3 es un factor de regulación clave en el control de la migración de las células precursoras miogénicas23,25.
El estudio de las bases celulares del proceso de cardiogénesis revela que son varios los tipos de células que contribuyen al crecimiento cardíaco. Las células del primer campo del corazón (FHF, siglas en inglés de first heart field) contribuyen únicamente a la formación del ventrículo derecho y del canal atrio-ventricular, mientras que las aurículas, ventrículo izquierdo y gran parte del tracto de salida, provienen de precursores mesenquimales que residen en el segundo campo del corazón (SHF, siglas en inglés de second heart field)17,18,20.
La vía de señalización celular Nodal/TGF tiene un importante efecto en los estadios tempranos de la diferenciación de las células embrionarias humanas pluripotenciales, que las dirigen a desarrollarse en diferentes linajes celulares. El gen SMAD3 o SEMA3D es un mensajero intracelular clave en la regulación de la vía de señalización Nodal/TGF, que tiene un importante papel en el desarrollo embrionario y particularmente, en el desarrollo del sistema cardiovascular15 26,27.
De igual forma, la vía de señalización Notch desempeña un papel fundamental en el desarrollo del período embrionario, pues es imprescindible para que las capas germinales den origen a los tejidos que constituyen un organismo multicelular. Esta vía de señalización determina, además, la especificación celular, la histogénesis y la morfogénesis de varios tejidos en el desarrollo embrionario de vertebrados e invertebrados, y resulta clave en la especificación de los miocardiocitos y los vasos sanguíneos. Esta vía está conservada evolutivamente en la escala filogenética y debe su nombre al fenotipo que causan mutaciones en el gen que codifica su receptor, el cual se caracteriza por escotaduras (Notch) en los márgenes alares de la Drosophila13,28.
EPÍLOGO
No existe una exacta correlación genotipo-fenotipo entre los mecanismos moleculares y los defectos morfológicos de las cardiopatías congénitas. Esto sucede porque muchas veces la formación adecuada de una estructura anatómica implica el correcto funcionamiento de varias vías de señalización celular que pueden involucrar el producto proteico de distintos genes. En este sentido, puede que en un mismo defecto congénito estén implicadas diferentes vías y mutaciones génicas, o por el contrario, que debido al efecto pleiotrópico de mutaciones, en uno de los genes críticos en el proceso de cardiogénesis, se originen diferentes tipos de cardiopatías congénitas; todo lo cual se resumirá en la segunda parte de este artículo donde se discutirán algunos aspectos sobre los genes y las vías de señalización implicados en el origen de las cardiopatías congénitas.

