










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Finlay
versión On-line ISSN 2221-2434
Rev. Finlay vol.6 no.3 Cienfuegos sep.-dic. 2016
PRESENTACIÓN DE CASO
Sarcoidosis: presentación de un caso y revisión de la literatura
Sarcoidosis: Case Presentation and Literature Review
Dayana Alomá Fortún , Javier Hernández Barrio , Viviana de la Concepción García Escudero
Hospital General Universitario Dr. Gustavo Aldereguía Lima, Cienfuegos, Cienfuegos, Cuba, CP: 55100
RESUMEN
La sarcoidosis es una enfermedad sistémica de etiología desconocida en la que se han implicado agentes infecciosos, polvos inorgánicos o sustancias orgánicas; caracterizada por la presencia de una inflamación granulomatosa no necrotizante con acumulación de linfocitos CD4+ y monocitos en los tejidos afectados. Se presenta el caso de un paciente que acudió al Hospital General Universitario Dr. Gustavo Aldereguía Lima de Cienfuegos refiriendo tos seca, febrícula, opresión torácica y ligera pérdida de peso de tres meses de evolución, que no cedían, a pesar de recibir tratamiento en su área de salud. Luego de realizarle varios estudios se diagnosticó una sarcoidosis pulmonar, enfermedad granulomatosa sistémica y proliferativa cuya etiología permanece en el anonimato. La biopsia continúa siendo la base para el diagnóstico definitivo. Por ser esta una entidad difícil de explicar y entender, además de ser escasamente diagnosticada en nuestro medio se decide presentar este caso clínico.
Palabras clave: sarcoidosis, informes de casos.
ABSTRACT
Sarcoidosis is a systemic disease of unknown etiology in which infectious agents have been implicated, inorganic powders or organic substances, characterized by the presence of necrotizing granulomatous inflammation with no accumulation of CD4 + lymphocytes and monocytes in the affected tissues. It is presented the case of a patient who went to the General University Hospital Dr. Gustavo Aldereguía Lima of Cienfuegos reporting dry cough, fever, chest tightness and slight weight loss three of three months evolution which did not improve despite receiving treatment in his health area. After several studies it was diagnosed a proliferative pulmonary sarcoidosis, systemic granulomatous disease whose etiology remains anonymous. Biopsy remains the basis for definitive diagnosis. As this is a difficult entity to explain and understand, besides being scarcely diagnosed in our area, it is decided to present that clinical case.
Key words: sarcoidosis, case reports.
INTRODUCCIÓN
De la sarcoidosis puede decirse con justicia que es un enigma envuelto en un misterio como lo califica Robbins.1 No solo se desconoce la etiología —aunque en la actualidad se afirma que es una enfermedad de origen inmune— sino que otros la consideran de causa desconocida.2 Muchos autores coinciden en definirla como una enfermedad sistémica de etiología desconocida en la que se han implicado agentes infecciosos, polvos inorgánicos o sustancias orgánicas, caracterizada por la presencia de una inflamación granulomatosa no necrotizante3 con acumulación de linfocitos CD4+ y monocitos en los tejidos afectados.4 La presentación y curso clínico varían desde la enfermedad asintomática con la resolución espontánea, hasta la falla multiorgánica y eventualmente la muerte,5 afectando órganos como: hígado, bazo, ojos, piel 6 y pulmón, este último en más del 90 % de pacientes. La sarcoidosis afecta a personas de ambos sexos, de diferentes razas y lugares geográficos, con predisposición entre los 20 y 40 años.7 El diagnóstico de sarcoidosis se basa en un cuadro clínico y radiográfico de presentación compatibles, así como en signos histológicos (granulomas no caseificantes en la biopsia, sin organismos o partículas). Varios informes han demostrado el valor de tomografía por emisión de positrones para detectar la enfermedad oculta que puede además ayudar a determinar un sitio accesible para la biopsia, en particular en los casos difíciles como el compromiso cardíaco o neurológico.8 Por ser esta una entidad difícil de explicar y entender, además de ser escasamente diagnosticada en nuestro medio se decide presentar ese caso clínico.
PRESENTACIÓN DEL CASO
Se presenta el caso de un paciente de 38 años, de color de piel negra, con antecedentes de asma bronquial desde temprana edad, que no precisaba tratamiento y que se dedicaba a los trabajos manuales en el arreglo de relojes con el uso de varios productos químicos. Este paciente acudió al cuerpo de guardia de medicina interna, refiriendo que desde hacía aproximadamente 2 meses venía presentando falta de aire, que se intensificaba a los esfuerzos, que se acompañaba de tos con expectoración amarillenta que se hacía más marcada por las noches y en las mañanas. Además refería opresión torácica de ligera intensidad y pérdida del apetito, palpitaciones y febrícula, razón por la cual acudió a su área de salud donde fue medicado con azitromicina durante 6 días, tratamiento con el cual no cedieron los síntomas. Se recibió en el Servicio con ligero empeoramiento de la clínica antes referida. Luego de proceder con el examen físico del paciente, a pesar de no obtener ningún dato clínico, se decidió indicar una radiografía de tórax, en la que llamaba la atención un marcado ensanchado simétrico bilateral en la región del mediastino. Se decidió ingresar con la impresión diagnóstica de neumopatía inflamatoria, y se indicó tratamiento antibiótico con cefuroxima 750 mg c/8 horas por 7 días. Luego del cumplimiento con la antibioticoterapia se realizó radiografía de tórax evolutiva cuyo informe por parte del departamento de radiología precisó mejoría radiológica, pero persistía el engrosamiento hiliar bilateral que se describe como hilios en patatas, dando como diagnóstico presuntivo el de sarcoidosis o linfoma, se sugirió además la realización de tomografía axial computarizada (TAC). Se realizaron además varios exámenes de hemoquímica, resultandos estos dentro de los valores normales. Se realizó TAC (simple y contrastada) donde se observaron múltiples adenopatías en todos los compartimentos del mediastino con acentuación de la trama reticular de ambos pulmones, lo cual realza aún más los diagnósticos anteriores. (Figuras 1, 2, 3 y 4). Se repitió la radiografía de tórax evolutiva a los 6 meses de tratamiento donde se evidenció la mejoría radiológica.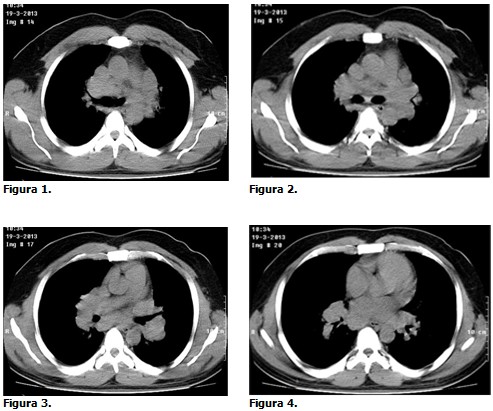
DISCUSIÓN
La sarcoidosis, (del griego sarx, que significa carne) o enfermedad de Besnier-Boeck, es un padecimiento granulomatoso sistémico, de carácter autoinmune,9-11 que en la mayoría de los casos remite espontáneamente aunque en ocasiones evoluciona hacia la cronicidad.12 El inicio del conocimiento de la sarcoidosis comienza en 1869, cuando Carl William Boeck, dermatólogo noruego, y Jonathan Hutchinson en 1877, célebre cirujano y dermatólogo inglés descubrieron, independientemente, ciertas lesiones dermatológicas raras.2 En su sinonimia guarda varios nombres: lupus pernio de Besnier, linfogranulamotosis benigna de Schaumann, síndrome o enfermedad de Jonathan Hutchinson, psoriasis papilar de Hutchinson, sarcoidosis subcutánea de Darier-Roussy, fiebre uveoparotídea de Heerfordt-Waldenström, osteítis cistoide múltiple de Jungling, tuberculosis magnocelular granulomatosa de Ziegler-Mylius-Schaumann, reticulosis epitelioide, sarcoide de Boeck, enfermedad del polen de pino, tuberculosis nodular, poliosteítis pseudoquística de Chevalier, sarcoidosis de Mortimer. 1,2 Su frecuencia varía en las diferentes regiones geográficas, y se informa una incidencia de entre 5 a 40 casos por cada 100 000 habitantes/año, siendo mayor en países escandinavos y en la población afro-norteamericana13-16 siendo ligeramente más común en mujeres. Latinoamérica es una región con baja incidencia, probablemente por diferencias genéticas y de exposición ambiental a determinados antígenos o por la falta de programas de relevamiento, y la alta prevalencia de otras enfermedades granulomatosas endémicas (tuberculosis, lepra, micosis profundas) que confunden el diagnóstico.17-20 En Cuba, el primer caso informado corresponde al municipio de Moa, en la provincia de Holguín, según informa Monier en 1991.21 Desde el punto de vista clínico puede afectar varios órganos:22 La radiografía de tórax puede mostrar signos de diferentes estadios: Estadio 1: linfadenopatía hiliar bilateral solamente (posibilidad de remisión espontánea 55-90 %). Estadio 2: linfadenopatía hiliar bilateral plus infiltrados pulmonares (40-70 %). Estadio 3: infiltrados pulmonares solos (10-20 %). Estadio 4: fibrosis pulmonar (0 %). Se observa una amplia variedad de anomalías de la piel, incluyendo lesiones maculopapular, lupus pernio (a menudo asociada con la enfermedad más crónica), nódulos e hiperpigmentación o hipopigmentación. El pronóstico de la enfermedad depende mucho de la forma clínica que adopte y la rapidez del diagnóstico. El caso presentado previamente solo presentaba afecciones a nivel pulmonar (Estadio 1), en el momento del diagnóstico y presentó muy buena respuesta al tratamiento con esteroides con evidente mejoría radiológica, por lo cual se puede inferir que su pronóstico será favorable con altas posibilidades de remisión.
REFERENCIAS BIBLIOGRÁFICAS
1. Reynolds HY. Sarcoidosis: impact of other illness on the presentation and management of multi-organ disease. Lung. 2002;180(5):281-99
2. Pila R. Sarcoidosis. La Habana: ECIMED; 2010
3. Weinberger SE. Sarcoidosis. En: Arend WP, Armitage JO, Clemmons DR, Drazen JM, Griggs RC, LaRusso. Cecil Medicine. 23 ed. Philadelphia: Saunders Elsevier; 2008: p. 931-2
4. Baughman RP, Lower EE. Sarcoidosis. En: Fauci AS, Kasper DL, Longo DL, Braunwald E, Hauser SL, Jameson JL, et al. Harrison´s Principles of Internal Medicine. 17 ed. New York: McGraw-Hill; 2008: p. 449-514
5. Iannuzzi M. Sarcoidosis. En: Goldman L, Schafer AI. Goldman´s. Cecil Medicine. 24 ed. Philadelphia: Elsevier Saunders; 2012: p. 582-5
6. Du Bois RM, Goh N, McGrath D, Cullinar P. Is there a role for microorganism in the pathogenesis of sarcoidosis?. J Int Med. 2003;253(1):4-17
7. Díaz JA. Sarcoidosis pediátrica. Revisión. Mediciego [revista en Internet]. 2013 [citado 12 Feb 2016];13 Suppl 1:[aprox. 10p]. Disponible en: http://bvs.sld.cu/revistas/mciego/vol13_supl1_07/revisiones/r5_v13_supl107.html
8. Iannuzzi M, Fontana JR. Sarcoidosis: clinical presentation, immunopathogenesis, and therapeutics. JAMA. 2011;305(4):391-9
9. González E, Vigliano C, Cáneva J. Sarcoidosis. Presentación clínica y pronóstico. Medicina. 2010;70(6):1-9
10. Baughman RP, Lower EE, du Bois RM. Sarcoidosis. Lancet. 2003;361(9363):1111-8
11. Nunes H, Bouvry D, Soler P, Valeyre D. Orphanet J Rare Dis. Sarcoidosis. 2007;2(1):46
12. Maña J. Sarcoidosis. 14 ed. Madrid: Ediciones Harcourt; 2000
13. Statement on sarcoidosis. Joint Statement of the American Thoracic Society (ATS), the European Respiratory Society (ERS) and the World Association of Sarcoidosis and Other Granulomatous Disorders (WASOG) adopted by the ATS Board of Directors and by the ERS Executive Committee, February 1999. Am J Respir Crit Care Med. 1999;160(2):736-55
14. Rybicki BA, Major M, Popovich J, Maliarik MJ, Iannuzzi MC. Racial differences in sarcoidosis incidence: a 5-year study in a health maintenance organization. Am J Epidemiol. 1997;145(3):234-41
15. Fernández E. Epidemiología de la sarcoidosis. Arch Bronconeumol. 2007;43(2):92-100
16. Baughman RP, Teirstein AS, Judson MA, Rossman MD, Yeager H Jr. Bresnitz. ACCESS Research Group. Clinical characteristics of patients in a case control study of sarcoidosis. Am J Respir Crit Care Med. 2001;164(1):1885-9
17. Pila R, Bestard A, Amador J, Boladeres C. Sarcoidosis: estudio de 30 pacientes. Rev Cuba Med. 1986;25(11):1027-37
18. Bethlem NM. Epidemiology of sarcoidosis in Brazil. Sarcoidosis. 1985;2(1):162
19. Silva LC, Corrêa da H, Felipe T, Cruz DB, Caraver F, Fernandes J, et al. Sarcoidose no sul do Brasil: Estudo de 92 pacientes. J Bras Pneumol. 2005;31(5):398-406
20. Purriel P, Navarrete E. Epidemiology of sarcoidosis in Uruguay and other countries of Latin America. Am Rev Respir Dis. 1961;84(5):155-61
21. Monier JP, Affre J, Bouskela R, Chermet J, Chevrot A, Coussement A, et al. Manual de radiodiagnóstico. La Habana: Editorial Científico-Técnica; 1991
22. Dempsey O, Paterson E, Kerr K, Denison A. ¿Pensó en Sarcoidosis? Revisión clínica. Intramed [revista en Internet]. 2009 [citado 25 Ene 2016]; . Disponible en: http://www.intramed.net/contenidover.asp?contenidoID=62457
Recibido: 09 de marzo de 2016.
Aprobado: 03 de julio de 2016.
Dayana Alomá Fortún. Especialista de I Grado en Medicina Interna. Profesor Instructor. Hospital General Universitario Dr. Gustavo Aldereguía Lima. Cienfuegos. Correo electrónico: dayana.aloma@gal.sld.cu

