










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
A escala mundial, las autoridades reguladoras son las responsables de garantizar que los productos farmacéuticos, incluyendo los de origen natural, sean eficaces y seguros (Agilent Technologies, 2017).
Las trazas de impurezas elementales inorgánicas pueden disminuir la estabilidad y acortar la vida de los fármacos, además de suponer un riesgo toxicológico por sí mismas y deben ser controladas atendiendo a las Buenas Prácticas en la Manufactura. Autoridades como la Farmacopea de Estados Unidos (United States Pharmacopeia, USP), el Consejo Internacional de Armonización de los requisitos técnicos para el registro de medicamentos de uso humano (International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use, ICH), han publicado en paralelo nuevas normas para el análisis individual de impurezas elementales en los productos farmacéuticos y sus ingredientes, lo cual ha impactado considerablemente a nivel regulatorio y en la industria farmacéutica. A esas nuevas normativas se han sumado otras autoridades como la Farmacopea Europea (European Pharmacopea, EuP) y la Farmacopea China (ChP), entre otros (USP, 2017; ICH, 2014; EuP, 2014; ChP, 2016).
En este sentido, incluyen una lista de 24 impurezas elementales, clasificadas según su toxicidad y probabilidad de ocurrencia y proponen una metodología basada en un análisis de riesgos que tendrá en cuenta la toxicidad de la impureza, la probabilidad de encontrarla en el fármaco y la dosis diaria máxima admisible para cada una, según la vía de administración (µg. día-1). Exigen el control de las impurezas inorgánicas más tóxicas y abundantes para el hombre, clasificadas como clase I (Cd, Pb, As y Hg) (USP, 2017; ICH, 2014) para todos los productos terminados y han definido límites máximos de Exposición Diaria Permitida (EDP), científicamente justificables (Teasdale, 2015; Agilent, 2017).
Además, introducen la aplicación de técnicas analíticas nuevas más sensibles, específicas, selectivas, precisas, seguras, con mayor reproducibilidad y mejor recuperación. Recomiendan el uso de técnicas instrumentales modernas, como la espectrometría de emisión óptica por plasma acoplado inductivamente (ICP-OES) o la espectrometría de masas acoplada a la espectrometría de emisión con fuente de plasma acoplado inductivamente (ICP-MS). Pueden utilizarse además técnicas alternativas, como la Espectrometría de Absorción Atómica (EAA), siempre que se demuestre que cumplen los requisitos de rendimiento definidos. Proponen igualmente el uso de recipientes cerrados en la digestión de muestras sólidas, con el fin de garantizar una recuperación cuantitativa de los elementos volátiles (USP, 2017; ICH, 2014).
Recomiendan cómo deben realizar los fabricantes la evaluación de riesgos de un producto farmacéutico para demostrar la conformidad con los límites establecidos por las normas. Entre las opciones posibles se incluyen el análisis directo del fármaco final, de cada uno de los componentes de la formulación, incluyendo el ingrediente activo, de las materias primas utilizadas, entre otras (USP, 2017; ICH, 2014).
Teniendo en cuenta que, la Directriz Q9 del ICH define la administración del riesgo como la aplicación sistemática de políticas, procedimientos y prácticas de las tareas de análisis, evaluación, control y monitoreo de la efectividad del control del riesgo y que la NC ISO/IEC 31000: 2018, plantea el proceso general de la gestión de riegos a través de cinco etapas: establecer el contexto, identificar los riesgos, analizar los riesgos, evaluar los riesgos y tratamiento de los riesgos, se pueden tomar estos criterios como base para realizar la evaluación de riesgos de impurezas elementales en los productos farmacéuticos, según requieren las normativas (ICH, 2005; NC ISO 31000, 2018).
En el Centro Nacional de Investigaciones Científicas de Cuba (CNIC) se producen principios activos de origen natural que han demostrado actividad antioxidante, los cuales son utilizados en la formulación de productos farmacéuticos y de suplementos. Como parte del control de calidad establecido, se realiza el análisis de impurezas elementales a los IA, teniendo en cuenta lo normado en farmacopeas e ICH.
El cambio absoluto de la USP e ICH en el enfoque para el control de las impurezas inorgánicas, al estimar nuevos límites para Cd, Pb, Hg y As, supone un impacto considerable en la calidad del producto, en las especificaciones, en el expediente de registro, en la analítica instrumental del laboratorio de control y en lo económico de la empresa productora en este sentido. Por tal motivo, el objetivo de este trabajo fue aplicar la evaluación de riesgos de impurezas elementales, conforme a USP e ICH, para ingredientes activos (IA) naturales producidos en el CNIC.
MATERIALES Y MÉTODOS
En este trabajo, se siguió la metodología general propuesta por las normativas de la USP farmacopea y las directrices Q3D del ICH para evaluar los riesgos potenciales por la presencia de impurezas elementales inorgánicas, en los productos farmacéuticos e ingredientes. La metodología consta de cinco etapas fundamentales (Figura 1):

Fig. 1 Metodología para la evaluación de riesgos potenciales por la presencia de impurezas elementales inorgánicas en productos farmacéuticos e ingredientes, según ICH y USP.
Para el desarrollo de la tercera etapa “Evaluación de Riesgos” se realizaron los siguientes pasos: un análisis del contexto, identificación de los riesgos, establecer el nivel del riesgo identificado sobre la base de la probabilidad de ocurrencia y de su posible impacto en los resultados del laboratorio, y por último, la decisión sobre el control del riesgo.
Análisis del contexto
El propósito del análisis del contexto y el establecimiento de criterios es adecuar el proceso de la gestión de riesgos para una evaluación eficaz y un tratamiento apropiado de los mismos.
El contexto del laboratorio se analizará de modo que permita identificar los factores externos e internos que puedan generar riesgo, impactando en los resultados del mismo, en este caso en el análisis seguro y valido de las impurezas inorgánicas en los ingredientes activos producidos en la empresa. En el contexto interno, debe tenerse en cuenta el direccionamiento estratégico y los objetivos del laboratorio, capacidades en términos de recursos y conocimientos: equipamiento, estándares de referencia, calidad de reactivos y materiales, métodos de ensayo, competencia del personal, normativas, condiciones ambientales, el aseguramiento metrológico, la información documentada, toma de decisiones, responsabilidades, etc. En el contexto externo, referido a aquel en el que se desarrolla el laboratorio y que le permite alcanzar sus objetivos, se puede incluir: ambiente social y cultural; percepciones y valores de las partes interesadas; requisitos legales, regulatorios, sociales, financieros, tecnológicos, económicos, medioambientales, etc. a nivel local, regional, nacional e internacional (Cox, 2021; Sammut-Bonnici y Galea, 2014).
Criterios para evaluar los riesgos
Los criterios a utilizar para la evaluación de los riesgos se establecerán teniendo en cuenta la probabilidad y el impacto. Para ello, se analizarán los requisitos normativos reglamentarios y legales, según USP e ICH, que puedan afectar el análisis de las impurezas elementales realizado por el laboratorio de Control de Calidad de EAA en los ingredientes activos producidos en la empresa. Los criterios se deben adaptar al propósito y al alcance específico del laboratorio. Considerar factores como la actividad en la que se puede presentar el riesgo, sus causas, la fuente de procedencia, consecuencias, forma en la que se expresarán las probabilidades, cómo se va a determinar el impacto, cómo se va a determinar el nivel del riesgo, las escalas de evaluación del impacto, de la probabilidad de ocurrencia, la valoración que se realice y la capacidad del laboratorio (ICH, 2005; NC ISO 31000, 2018; Resolución 60, 2011).
RESULTADOS Y DISCUSIÓN
Identificación de impurezas elementales, toxicidad y límites de exposición diaria permitida (EDP)
La lista de elementos inorgánicos, clasificados por las nuevas directivas en tres grupos (clase 1, clase 2A, clase 2B y clase 3), tiene en cuenta la toxicidad de los elementos y la probabilidad de que estén presentes en los productos farmacéuticos. Como se mencionó anteriormente, los elementos de clase 1(Cd, Pb, As y Hg) más tóxicos y abundantes deben incluirse en la evaluación de riesgos de todos los productos farmacéuticos.
Se revisó y documentó la información toxicológica de cada una de las principales impurezas elementales a monitorear, de manera obligatoria. El valor de EDP (Exposición Diaria Permitida) para cada impureza, según la vía de administración oral (µg/día), se encuentra homologado tanto en las guías ICH como en la USP. La información se encuentra resumida en la Tabla 1. Se destaca que los valores de exposición diaria máxima permisible para la administración de productos en (μg/día) están basados en una persona promedio de 50 kg.
Tabla 1 Información toxicológica de las impurezas elementales a considerar en el análisis de riesgo y el valor de EDP.

La toxicidad potencial de una impureza elemental varía en función de la vía de exposición. Por tal razón, las impurezas elementales deben incluirse en una evaluación de riesgos del producto que resulte adecuada para la vía prevista de administración del producto farmacéutico final. Conforme a las normativas, la presencia de los elementos de la clase 1 tiene que considerarse en la evaluación de riesgos para todos los tipos de fármacos. Por el contrario, la presencia de un elemento de clase 3 solo será necesaria tenerla en consideración si se trata de un fármaco que se administra por vía parenteral o de inhalación.
Tanto USP 232 como ICH Q3D acuerdan que la EDP se aplica solo al fármaco. Sin embargo, existen límites internos que se pueden establecer de acuerdo con su evaluación de riesgos, según la contribución de cada componente a la impureza elemental final contenida en el fármaco.
Identificación de fuentes potenciales de impurezas elementales
Origen de las impurezas inorgánicas en los IA naturales
Las nuevas directrices Q3D del ICH y el nuevo capítulo 232 de la USP incluyen elementos catalizadores y otros contaminantes inorgánicos que pueden llegar a los productos farmacéuticos a través de las materias primas, el proceso de fabricación, el entorno, los materiales de acondicionamiento y los sistemas de envase y cierre. Con el enfoque de evaluación de riesgos, se evalúa la contribución de impurezas de cada componente en el producto terminado. Este enfoque permite a los fabricantes proporcionar información crucial sobre la contribución de las impurezas en los productos farmacéuticos finales a partir de excipientes o ingredientes farmacéuticos activos.
En cualquier caso, es necesario identificar y documentar el origen de las posibles fuentes de impurezas elementales en el proceso de fabricación del IA a estudiar para asegurar que el producto cumpla el requerimiento regulatorio.
Como se aprecia en la Figura 2, el origen de las impurezas elementales en los IA naturales producidos en la empresa puede provenir, entre otros, de contaminantes ambientales procedentes de aguas de vertimiento, fumigaciones, solventes que se utilizan en la etapa de extracción del proceso productivo, interacción con los equipos de fabricación y las materias primas de origen natural a partir de las cuales se obtienen los IA como son, la cera de la caña de azúcar, la cera de abeja y el fruto de la palma real.

Evaluación de riesgos
Análisis del contexto
El personal del laboratorio cuenta con ventajas en el ámbito preventivo y también de la seguridad ya que tiene conocimiento sobre la existencia de peligros y de los principales riesgos para la calidad de sus resultados, la salud y el medio ambiente. El laboratorio dispone de una serie de instalaciones y servicios generales de gas, agua, aire comprimido, electricidad, etc., que se hallan en buen estado y están sometidas a un mantenimiento adecuado para garantizar un riesgo nulo o escaso de provocar daños a la calidad del resultado analítico y a la salud del personal.
En el análisis de contexto, se tuvo en cuenta la instalación, las condiciones ambientales, los reactivos y materiales de referencia utilizados, las técnicas analíticas que se utilizan, las características de las muestras, las condiciones de limpieza y cuidado de la cristalería, las regulaciones, las legislaciones, las partes interesadas, el entorno, etc. Se utilizó una tormenta de ideas, se trabajó en grupo, se realizaron entrevistas a expertos y se revisaron guías para la identificación de riesgos. También fueron revisados datos históricos de resultados de ensayos realizados por el laboratorio de EAA.
Además, fue motivo de análisis el hecho de que los procedimientos utilizados para la determinación de los contenidos de Cd, Pb, As y Hg, son procedimientos desarrollados en el laboratorio de Control de Calidad de EAA del CNIC. En el caso del cadmio y el plomo, se efectúa una digestión en sistema abierto y la determinación se realiza por absorción atómica con llama aire-acetileno. Para el mercurio se utiliza un sistema cerrado de digestión (bombas Para) y la cuantificación se lleva a cabo por la técnica de vapor frio. El arsénico se determina por generación de hidruros, previa digestión ácida de la muestra en sistema abierto.
Todos los procedimientos analíticos se encuentran debidamente documentados, implementados y validados. Los instrumentos de medición que se utilizan en el laboratorio para llevar a cabo los procedimientos están calificados y calibrados. Los patrones de referencia que se utilizan son de alta pureza, confiables y certificados.
Aun cuando las técnicas analíticas de control utilizadas en el laboratorio de Control de Calidad de EAA del CNIC no son las recomendadas por farmacopeas, las mismas son específicas, precisas, exactas y adecuadas para el propósito. Además, han demostrado aplicabilidad a los productos de interés.
Criterios para evaluar los riesgos
En la Tabla 2 se muestran los criterios para la evaluación del riesgo, teniendo en cuenta la probabilidad de ocurrencia y el impacto (ICH Q9, 2005).

Identificación de riesgos
Los riesgos se identificaron en el laboratorio a partir del conocimiento de los procesos técnicos e investigativos que en él se realizan, así como del análisis de contexto.
Como resultado del análisis anterior, se identificaron dos riesgos: el primero, la calidad del producto por la posible presencia de impurezas elementales inorgánicas en los IA y el segundo, la posible insuficiencia de las técnicas analíticas de control con que son determinadas las impurezas elementales en el laboratorio.
Evaluación del riesgo
A pesar de la existencia de diversos métodos de evaluación de riesgos, los más utilizados son los métodos cualitativos. En todos los casos se ha de profundizar en dos aspectos claves, probabilidad y consecuencia. Es necesario el entendimiento del riesgo obtenido durante el análisis para tomar decisiones sobre las acciones futuras.
Posterior a la identificación, se realizó un análisis de los riesgos, la naturaleza de estos, sus características, las causas asociadas a ellos para determinar la probabilidad de ocurrencia y las posibles consecuencias o impacto sobre los resultados del laboratorio, así como para la continuidad del proceso de determinación de las impurezas elementales Cd, Pb, As y Hg en él. El análisis tuvo como propósito, además, determinar los controles existentes. En este sentido, el laboratorio tuvo en consideración el aporte de impurezas elementales que tienen su origen en las materias primas que se emplean en las producciones de IA, para lo cual tiene establecido un control de cuantificación de IE en las mismas.
Para el análisis, se tuvieron en cuenta los criterios de evaluación del nivel de riesgo (Tabla II), según probabilidad e impacto (ISO 14971:2007, ONN, 2018). Se analizaron detalladamente experiencias anteriores del propio laboratorio, opiniones de expertos, experiencias de otros laboratorios, incertidumbres, fuentes de riesgo, eventos, magnitud de las consecuencias, probabilidades, escenarios, controles existentes y su eficacia, los niveles de sensibilidad y de confianza de la información y los recursos disponibles. Los riesgos identificados fueron evaluados de acuerdo a una escala de 1, 2 y 3 mediante la multiplicación de la probabilidad por el impacto. Este resultado no es más que el nivel del riesgo y se resume en la Tabla 3.
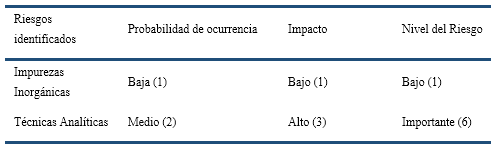
Como se aprecia en la Tabla 3, el riesgo a la calidad del producto por la posible presencia de impurezas elementales en el IA es bajo, resultado relevante teniendo en consideración el impacto a nivel regulatorio que tuvo el cambio absoluto de la ICH y la USP para el control de este tipo de impurezas en la industria farmacéutica. Sin embargo, el riesgo debido a la posible insuficiencia de las técnicas analíticas empleadas en el laboratorio para el control de las impurezas es importante. Esto implica un desafío, considerando que la USP y la ICH introdujeron también un cambio significativo en el campo analítico, con respecto al uso de nuevas técnicas instrumentales de análisis para el control de impurezas elementales.
En esta etapa de evaluación, comparar el nivel del riesgo con los criterios que fueron adoptados permitió considerar y priorizar el tratamiento a seguir con los riesgos identificados. Sobre la base de los resultados de la evaluación se decidió: si eran necesarias acciones adicionales, no tomar acciones, considerar opciones para el tratamiento del riesgo o mantener los controles existentes. De esta forma, la evaluación de los riesgos se aborda como un control preventivo para minimizar el resultado no deseado, es decir, la presencia de impurezas elementales en los IA producidos.
Valoración del límite de la concentración diaria apropiada para cada impureza en el producto, respecto al límite de detección (LD) del método
Las nuevas normativas plantean el requerimiento del cálculo de los límites de la concentración diaria apropiada para cada impureza en un producto (calculados según la ecuación 1), teniendo siempre en cuenta los valores de Exposición Diaria Permitida (basados en una dosis diaria máxima supuesta de 10 g/día) y la dosis diaria máxima.

(Ec. 1)
Como se observa en la Tabla 4, los LD de los métodos analíticos implementados por el laboratorio de Control de Calidad de EAA del CNIC resultan inferiores a los límites de concentración apropiados calculados. Por lo cual, resultan adecuados para la cuantificación de las impurezas elementales de manera confiable a los niveles requeridos, según ICH y USP.

Estrategia de control en función de los valores/límites establecidos. Determinación de acciones.
En esta etapa se verificó si los riesgos están bajo control, la aplicación de las medidas de prevención establecidas, así como su efectividad. Para ello, se tuvo en cuenta la estrategia de control propuesta por el ICH (Tabla 5).

A partir del análisis realizado, y de los resultados de la identificación y evaluación de riesgos ejecutada en el laboratorio de control de EAA se demostró que los productos cumplen con el atributo de calidad establecido para el contenido de impurezas elementales a los niveles establecidos por las guías ICH y normas de la USP. Esto implica que, en este sentido, los IA pueden utilizarse en la fabricación de productos comercializables y cumplen con los más altos estándares de calidad.
Si bien las guías están destinadas en última instancia a centrarse en la calidad del producto farmacéutico final, la evaluación del riesgo real toca todas las facetas de su fabricación y queda a elección del productor la opción para determinar el contenido de impurezas elementales, en el fármaco final, como sumatoria de cada materia prima utilizada en la formulación o de los componentes individuales, etc., para demostrar la conformidad con los límites establecidos por las nuevas normas.
Conforme con lo recomendado por las nuevas normativas del ICH y la USP para la estrategia de control, la acción a realizar es documentar todo el proceso de evaluación de riesgos, informar y conservar la información documentada, sin necesidad de acciones adicionales. La información relacionada fue aprobada por las personas que toman las decisiones y se puso en conocimiento de las partes interesadas.
Otros autores han investigado la presencia de impurezas elementales clase I en productos farmacéuticos con un enfoque a riesgos. Por ejemplo, Kelly y Lewen (2014), discuten una estrategia para integrar la evaluación toxicológica de las impurezas elementales encontradas durante el análisis de extraíbles con las guías de la Farmacopea de los Estados Unidos, la Farmacopea Europea y la Agencia Europea de Medicamentos, para desarrollar un enfoque basado en el riesgo para el análisis de impurezas elementales en estudios de lixiviables.
Ariza (2016) por su parte, presenta un estudio del control y análisis de la posible presencia de impurezas elementales en principios farmacéuticos activos conforme a la USP 232 y 233, para asegurar que los mismos cumplan con los requisitos de seguridad y con las características de calidad y pureza que les son especificadas. Este autor desarrolló y validó métodos analíticos generales mediante la técnica de ICP-MS para la cuantificación de IE Clase 1, Clase 2A y Clase 2B para los productos desarrollados y producidos en la empresa farmoquímica. Los resultados permitieron comprobar la funcionalidad de este tipo de técnica para la cuantificación de IE en las diferentes etapas del proceso. Para determinar las etapas críticas del proceso aplicó herramientas como el manejo del riesgo a la calidad y espacios de diseño para un mejor control de las pruebas a realizar.
Otros autores reportan la detección y cuantificación de las impurezas inorgánicas más toxicas y abundantes como Cd, Pb, As y Hg por EAA, abordándolos con otros enfoques, tanto en productos farmacéuticos como en productos fitoterapéuticos, herbarios, ayurvedicos, alimentos, y plantas medicinales, entre ellos destacan Madueño Ventura (2017), Urtecho (2020), Torres Vásquez (2020) y Ruiz Pérez (2019). Ellos demuestran que es posible utilizar procedimientos alternativos, siempre que se demuestre que cumplen con los requisitos de rendimiento definidos en los capítulos correspondientes de las farmacopeas.
A partir de todo el análisis anterior, y de los resultados de la identificación y evaluación de riesgos realizada en el Laboratorio de Control de EAA, donde se determinan las impurezas elementales inorgánicas que pueden estar presentes en los ingredientes activos producidos en el CNIC, se hace necesario considerar el control actual de impurezas elementales en IA, ante nuevos requerimientos de farmacopea en productos terminados; mantener el control establecido para las materias primas en la producción de los mismos; revisar y replantear los límites de impurezas elementales a partir de los nuevos límites establecidos por USP e ICH y el cálculo de los límites de concentración diaria adecuados. Además, teniendo en cuenta que el riesgo identificado por la posible insuficiencia de las técnicas analíticas empleadas para el control de las impurezas elementales en el laboratorio fue evaluado como Importante, se hace necesario un seguimiento y valoración de mejora del equipamiento analítico de control.
CONCLUSIONES
Se realizó la Evaluación de Riesgo de impurezas elementales inorgánicas Clase I (Cd, Pb, As y Hg) en principios activos de origen natural producidos en el CNIC, teniendo en cuenta los nuevos criterios, enfoques y límites establecidos por la ICH y la USP y el análisis de riesgo obligatorio de impurezas de clase I en todos los productos farmacéuticos.
La identificación y evaluación de riesgos demostró que el riesgo asociado a la presencia de impurezas elementales es bajo, lo cual evidencia la eficacia de los controles implementados.
El enfoque basado en el riesgo, combinado con el conocimiento y control de las fuentes potenciales de IE en la producción de IA, permitirá mantener una estrategia de control eficiente para estas impurezas en el CNIC.

