










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Química
versión On-line ISSN 2224-5421
Rev Cub Quim vol.30 no.1 Santiago de Cuba ene.-abr. 2018
ARTICULOS
Cuantificación de los contenidos de nitrato en aguas de pozo y aguas residuales con el empleo de electrodos selectivos
Quantification of nitrate contents in waters and wastewater with the use of selective electrodes
MSc. Surey Ramírez-GonzálezI, Lic. Rujaime Cruzata-ManuelII, Dra. C. Margie Zorrilla-VelazcoI, Ing. Yamell Jiménez-PrietoI, Dra. C. Maira María Pérez-VillarI, Lic. Yennier Cruz-BermúdezII, Lic. Luis Ernesto Morera-HernándezII
ICentro de Estudio de Química Aplicada, Universidad Central de Las Villas, Cuba, sureyrg@uclv.edu.cu, margiezv@uclv.edu.cu, yamelljp@uclv.edu.cu, mairapv@uclv.edu.cu
IIDepartamento de Licenciatura en Química, Universidad Central de Las Villas, Cuba, yennierc@uclv.edu.cu
RESUMEN
En este trabajo se realiza la validación parcial del método potenciométrico empleando electrodos selectivos, para la cuantificación del contenido de nitrato en muestras de aguas de pozo y residuales. Se evalúan los parámetros de desempeño: linealidad, sensibilidad, límite de detección (LD), límite de cuantificación (LC), precisión y efecto matriz. Se obtuvo un intervalo lineal entre 0,5 y 100 mg/L, un límite de detección de 0,367 mg/L y de cuantificación de 1,224 mg/L. El coeficiente de variación se encuentra por debajo de dos, siendo mucho menor a concentraciones de nitrato por encima de 30 mg/L. Se llevó a cabo además una comparación entre la curva de calibración establecida y el método de adición del patrón, observándose efecto matriz. Se realizó la determinación de nitrato en muestras de agua de pozo y aguas residuales, no cumpliendo las mismas con las normas cubanas en materia de agua y aguas residuales.
Palabras clave: nitrato, aguas de pozo, aguas residuales, electrodo selectivo, validación.
ABSTRACT
The partial validation of the potentiometric method was conducted by means of selective electrodes for the nitrate contents quantification in samples of groundwater and wastewaters. First, the performance parameters were evaluated: linearity, sensitivity, limit of detection (LD), limit of quantification (LC), precision and matrix effect. A linear range was obtained from 0,5 to 100 mg/L of nitrate, detection limit of 0,367 mg/L and the quantification of 1,224 mg/L. The coefficient of variation is below two, which is considerably lower at nitrate concentrations above 30 mg/L. A comparison was also made between the established calibration curve and the standard addition method, displaying the matrix effect. The determination of nitrate was performed in samples of drinking water and wastewater, and did not comply with Cuban water and wastewater standards.
Keywords: nitrate, water supply, wastewaters, selective electrode, validation.
INTRODUCCIÓN
En la evaluación de la calidad del agua los compuestos de nitrógeno presentan una gran importancia debido a su participación en los procesos biológicos que ocurren en ella, como también por la toxicidad que pueden presentar para los organismos acuáticos. Particularmente, en la calidad del agua de abasto reviste gran importancia la determinación del ión nitrato, el cual se encuentra presente de forma natural tanto en aguas superficiales como subterráneas en bajas concentraciones, pero el consumo de agua con alta concentración del mismo puede causar deficiencia en el transporte de oxígeno en la corriente sanguínea. La contaminación del agua subterránea por nitratos producto de la fertilización excesiva, práctica agrícola o inadecuada disposición de las aguas residuales, tanto industriales como domésticas constituye un problema de actualidad. Las consecuencias asociadas a este deterioro pueden repercutir sobre la salud de las comunidades a corto, mediano o largo plazo [1-3].
En este sentido, el desarrollo de técnicas de análisis físico-químico para este contaminante en las aguas constituye una herramienta para su monitoreo y control, evitando el consumo de agua contaminada por la población. La dificultad de la determinación de nitratos en aguas está dada por la gran posibilidad de encontrar sustancias interferentes, amplios intervalos de concentración y la complejidad que presentan las diferentes técnicas analíticas, por lo que la validación de métodos de ensayos constituye una garantía de la calidad de los resultados que se reportan [4].
MATERIALES Y MÉTODOS
Se utilizaron reactivos de grado analítico y agua desionizada (ultrapura) con una conductividad por debajo de 2 µS/cm para todos los ensayos. Se empleó un electrodo de ión selectivo (ISE) a iones nitrato (NO3-) NO 501 (marca: WTW) y un Electrodo (ISE) (NO3-) R 503 (Referencia) (marca: WTW) con solución de llenado (ELY/BR/503/N) (solución de (NH4)2SO4). Para las medidas se utilizó un potenciómetro inoLab pH ION 735 (WTW). Como solución patrón se empleó NaNO3 (ES/NO3-) y para el ajuste de la fuerza iónica un Buffer (TiSAB/NO3) (solución de Al2(SO4)3). La cristalería utilizada en las determinaciones, está debidamente verificada por la Oficina Territorial de Normalización (OTN) en Villa Clara, Cuba.
Criterios de verificación aplicados al método desarrollado
La verificación del método se llevó a cabo según lo establecido en las normas de validación [9-11]. Se evaluaron los siguientes parámetros de verificación: especificidad, intervalo lineal, límite de detección, límite de cuantificación, precisión y efecto matriz. Todos los cálculos realizados durante el procesamiento de los datos experimentales se realizaron con el programa Microsoft Office Excel 2010.
Intervalo de respuesta lineal
Se realizaron tres réplicas de curvas de calibración (serie A, serie B y serie C) con concentraciones del patrón de NaNO3 comprendido entre 0,5 y 100 mg/L y solución de ajuste de fuerza iónica 1, 3 y 5 mL respectivamente. Cada punto de las tres curvas se leyó 10 veces y luego se construyó la curva promedio de cada serie de datos. La linealidad de los datos se evaluó a través de los test de Tiley [9], Falta de Ajuste (LOF) [12, 13], análisis del Coeficiente de Calidad (QC) [13, 14] y de los residuos de la regresión lineal [9].
Límite de detección y límite de cuantificación
Se determinó el límite de detección y de cuantificación de acuerdo con la definición recomendada por la IUPAC [15]. Para ello se prepararon diez réplicas de soluciones blanco para cada ión y se realizó la medición de los valores de potencial.
Precisión
Repetibilidad: Se realizó la lectura de las soluciones patrón de nitrato en tres niveles de concentración diferentes, comprendidos dentro del rango de estudio (n=10).
Las soluciones fueron preparadas en un mismo día y con el mismo analista. Se calculó la media de la concentración, la desviación estándar y el coeficiente de variación de la repetibilidad (CVr) de los resultados obtenidos.
Precisión intermedia: Se realizó en condiciones de repetibilidad pero en tres días diferentes y con diferentes analistas. Los valores obtenidos se analizaron en un solo grupo y se calculó la media (Xm); la desviación estándar (Sb) y el coeficiente de variación (CV %) de las determinaciones efectuadas.
Efecto matriz
Se llevaron a cabo estudios de comparación de dos funciones de calibración [12], una con patrones y otra obtenida por adición de estándar, para todas las muestras analizadas. Se comparan las varianzas de los calibrados mediante un test de F de Snedecor [12]. Para este test se consideran las siguientes hipótesis: H0: Varianzas iguales =; Fexp < Fcrítico y H1: Varianzas distintas ?; Fexp = F crítico.
Se realiza un t-Student [7]. Las hipótesis a tener en cuenta son las siguientes: H0: Pendientes iguales b1 = b2; t exp<t crítico y H1: Pendientes distintas b1 ? b2; t exp = t crítico. Y por último, se comparan los términos independientes para los que se considera H0: ordenadas iguales a1 = a2; z exp < z crítico y H1: ordenadas distintas a1 ? a2; z exp = z crítico.
RESULTADOS Y DISCUSIÓN
Linealidad
Las curvas de calibración para la determinación de nitrato de cada serie, luego de eliminar los datos anómalos, se muestran en la figura 1.
Los resultados de los test de linealidad realizados a los datos se muestran en la tabla 1, obteniéndose que para el test de Tiley Fcalculado< Fcrítico, lo que reporta la existencia de linealidad en las tres curvas de calibración según establece [11].
El mismo comportamiento se observó para el análisis del coeficiente de calidad, pues en todos los casos presenta un valor menor que el 3 % [15].
En los resultados del test de falta de ajuste, se encontró que Fexp < Fcrítico para la serie A. La varianza de falta de ajuste no será estadísticamente mayor que la del error puro, por lo que no existe un error debido a la falta de ajuste del modelo que no pueda ser atribuido al propiamente aleatorio, por lo tanto, el modelo lineal se ajusta perfectamente a los datos experimentales. Sin embargo, para la Serie B y Serie C, Fexp > Fcrítico; lo que indica que los valores de estas curvas de calibración no se ajustan al modelo lineal seleccionado [13]. El coeficiente de correlación de la curva de calibración de la serie A y serie B indica que existe una correlación aceptable entre las variables analizadas, al ser mayor que el valor crítico reportado; no siendo así para el caso de la curva de calibración de la serie C, dónde se evidencia la falta de correlación, ya que rexp (0,998) < rcrítico (0,999) criterio de aceptación establecido en [11] para este parámetro.
TABLA 1. VALORES DE LOS TEST DE LINEALIDAD REALIZADA A LAS CURVAS DE CALIBRACIÓN
| Tiley | LOF | OC | R | |||||
| Calculado | Crítico | Calculado | Crítico | Calculado | Crítico | Calculado | Crítico | |
| Serie A | 0,000 524 | 1,432 9 | 1,338 775 | 2,129 4 | 0,16 % | 3 % | 0,999 753 | r>0,999 |
| Serie B | 0,002 172 | 29,252 31 | 0,31 % | 0,999 019 | ||||
| Serie C | 0,000 327 | 1,429 8 | 30,519 43 | 2,127 8 | 0,46 % | 0,997 882 | ||
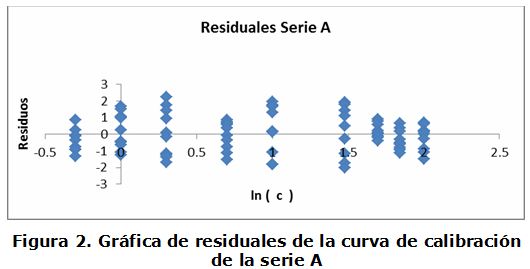
En las series B y C se observa una pérdida progresiva de la linealidad, lo que puede ser debido a la elevación del porciento de solución de ajuste de fuerza iónica añadido a estos patrones, con relación a los añadidos a la serie A [17]. La evaluación de la condición de homocedasticidad se realizó mediante el análisis de los residuos (figura 2), observándose la distribución homogénea de los mismos, por lo que se considera que el modelo presenta un buen ajuste a los datos [18]. Estos resultados evidencian la linealidad de los datos obtenidos de la serie A, por lo que el análisis del resto de los parámetros de desempeño del método se realizó sobre la curva de calibración de la serie A.
Sensibilidad
El valor de la pendiente de esta ecuación es igual 0,128 3; el cual se corresponde con el valor del coeficiente angular (Φ). Se calcula la sensibilidad del método a partir del intercepto de la ecuación de regresión ajustada para la curva logarítmica y la misma tiene un valor de 0,019 2 lo que indica que el método es sensible y que pequeñas variaciones en la concentración producen grandes cambios en la diferencia de potencial.
Límite de detección y límite de cuantificación
Los valores del límite de detección y el límite de cuantificación para el método de determinación de iones nitrato se muestran en la tabla 2. El límite de detección obtenido es inferior al reportado por Murray et al. y Wang, Q. et al. [19, 20].
TABLA 2. VALORES DE LOS LÍMITES DE DETECCIÓN Y CUANTIFICACIÓN PARA EL MÉTODO EN ESTUDIO
| c(NO3) mg/L | |
| Límite de Detección=3*SD | 0,367 |
| Límite de Cuantificación=10*SD | 1,224 |
Precisión
Repetibilidad
En la tabla 3 se muestran los resultados de la concentración de nitrato, obtenidos para el estudio de repetibilidad sobre la base de 20 determinaciones realizadas en tres niveles de concentración: 0,5 (bajo); 50,0 (medio) y 100,0 (alto) mg/L; bajo condición de repetibilidad. Se determina el coeficiente de variación, obteniendo valores menores que el 11 % para los tres niveles analizados, lo que se corresponde con el criterio de aceptación utilizado para este parámetro [11].
TABLA 3. VALORES DE CONCENTRACIÓN DE NITRATO EN MUESTRAS PATRONES EN CONDICIONES DE REPETIBILIDAD
| Parámetro | Nivel bajo (0,5 mg/L) | Nivel medio | Nivel Alto (100,0 mg/L) |
| Promedio | 0,50 | 49,55 | 101,30 |
| SD | 0,007 | 0,936 | 2,670 |
| C.V Repetibilidad (%) | 1,49 | 1,89 | 2,64 |
Precisión intermedia
Los resultados de la concentración de nitrato, obtenidos para el estudio de reproducibilidad (tabla 4), muestran que el coeficiente de variación (%) para cada día fue menor que 11 %, lo que indica que hay buena reproducibilidad en los análisis [21, 22].
TABLA 4. VALORES DE UNA MUESTRA ESTÁNDAR EN DIFERENTES DÍAS POR DISTINTOS ANALISTAS
| Media | 0,50 | 0,51 | 0,50 |
| Desviación Estándar (SD) | 0,008 | 0,006 | 0,009 |
| Coeficiente de Variación (%) | 1,51 | 1,26 | 1,79 |
Los cálculos realizados para los coeficientes de variación entre los días y de Horwitz [23], confirman que CV % entre días/analistas (1,054) << CV % Horwitz (11 %), por tanto se considera este criterio como de precisión aceptable para el método. La comparación por ANOVA de los valores de F obtenidos evidencian que Fexp (2,317) < Fcrítico (3,68). Criterio que demuestra que los resultados obtenidos en los diferentes días y utilizando analistas diferentes, no difieren entre sí y que el método es preciso.
La comparación del valor de la C de Cochram calculado con el tabulado, muestra que C calculado (0,488) < C tabulado (0,707); por lo que la variación del método se considera igual para los tres días y los analistas bajo estudio.
Como el estudio de la precisión, evaluado a través de los resultados de los ensayos de repetibilidad y precisión intermedia, cumple con los criterios de aceptación establecidos, se considera que el método estudiado es capaz de dar resultados semejantes de concentración de nitrato en agua cuando se aplica varias veces a una muestra.
Efecto matriz
Se realizó un test de comparación de dos rectas, para conocer si las curvas de calibración con patrones y con adición de estándar difieren significativamente. Se comprobó la igualdad de las varianzas de ambos calibrados mediante la prueba F de Fisher, seguidamente se analizó la igualdad de las pendientes, mediante el estadístico t-Student. De forma análoga se comprobó la igualdad de los términos independientes (tabla 5).
TABLA 5. PARÁMETROS ESTADÍSTICOS DEL ESTUDIO DE COMPARACIÓN DE LAS CURVAS DE CALIBRACIÓN
| Muestras | Pendientes | Términos independientes | ||||||
| Fexp | Fcrítico | texp | t | Fexp | Fcrítico | texp | tcrítica | |
| Pozo 1 | 4,191 5 | 2,029 8 | 4 720,32 | 2,12 | 0,024 1 | 0,573 8 | 28,815 8 | 1,98 |
| Pozo 2 | 4,822 2 | 6 903,31 | 0,025 4 | 24,163 1 | ||||
| Pozo 2 | 2,508 1 | 3 861,99 | 0,023 3 | 24,372 | ||||
| Fertilizantes | 6,688 7 | 12 861,19 | 0,060 8 | 4,325 3 | ||||
| Saladero de pieles | 4,442 2 | 5 599,47 | 0,024 5 | 39,924 3 | ||||
Como F calculado es mayor que F crítico (contraste de dos colas) para todas las muestras analizadas, la hipótesis alternativa se toma como cierta y las varianzas de los calibrados difieren significativamente. [24]. Se comprobó además que el estadístico t calculado resultó mayor que el valor tabulado, lo que permite aceptar la hipótesis alternativa, demostrando que las pendientes son estadísticamente diferentes.
Para los términos independientes los valores de F obtenidos de la comparación de las varianzas de los residuos del análisis de regresión realizado a los calibrados con patrones y con adición de estándar sobre las muestras reales, muestran que Fexp < Fcrítico (varianzas estadísticamente iguales), por lo que se acepta la hipótesis nula. El estadístico t-Student calculado presenta valores para los cuales se cumple que texp = t rítico. Se acepta la hipótesis alternativa, concluyéndose que los términos independientes son estadísticamente diferentes. Se demostró que las pendientes de los calibrados con patrones y con adición de patrón sobre muestras reales son estadísticamente diferentes, lo que permite afirmar que existe efecto matriz en las muestras analizadas.
Los resultados de concentración de nitrato obtenidos en muestras de agua de diferente procedencia se presentan en la tabla 6, observándose que las aguas de pozo analizadas no cumplen con los requisitos para consumo humano, observándose una concentración de nitrato en las muestras de agua de pozo por encima del Límite Máximo Admisible (LMA), según NC: 827/2010 [25]. Estos resultados pueden deberse a la cercanía de dichos pozos a grandes asentamientos poblacionales con ausencia de alcantarillado. En el caso de las aguas residuales también son elevados los valores de concentración de nitratos lo que puede estar relacionado con la presencia de compuestos nitrogenados en los procesos.
TABLA 6. CONCENTRACIÓN DE NITRATO EN MUESTRAS DE AGUA DE POZO Y AGUAS RESIDUALES
| Muestras | CNO3-(mg/L) | LMA según NC 827/2010 |
| Pozo 1 | 141,8 ±0,88 | 45 |
| Pozo 2 | 82,1±0,15 | 45 |
| Pozo 3 | 362,5±0,41 | 45 |
| Aguas residuales de la fábrica de fertilizantes | 261,9±0,23 | - |
| Aguas residuales de saladero de pieles | 246,8±0,28 | - |
CONCLUSIONES
El método de determinación de nitrato mediante electrodos selectivos cumple con los parámetros de desempeño linealidad, sensibilidad, límite de detección, límite de cuantificación y precisión, demostrándose que se puede emplear para la determinación de este parámetro en aguas de pozo y aguas residuales. Se comprobó la existencia de efecto matriz sobre las muestras analizadas. El estudio comparativo de los análisis realizados en muestras de aguas de pozo con respecto a las normas cubanas, arrojó que no cumplen con este parámetro por lo que no están aptas para el consumo humano.
REFERENCIAS BIBLIOGRÁFICAS
1. BEUTEL, M.; DUVIL, R. ; CUBAS, F.; MATTHEWS, D.; WILHELM, F.; GRIZZARD, T.; AUSTIN, D.; HORNE , A.; GEBREMARIAM, S. "A Review of Managed Nitrate Addition to Enhance Surface Water Quality". Critical Reviews in Environmental Science and Technology. 2016, 46(7), 673-700.
2. CAMARGO, J. A.; ALONSO, A. "Ecological and toxicological effects of inorganic nitrogen pollution in aquatic ecosystems: A global assessment". Environment International. 2006, 32(6), 831-849.
3. ALMASRI, M.; KALUARACHCHI, J. "Assessment and management of long-term nitrate pollution of ground water in agriculture-dominated watersheds". Journal of Hydrology. 2004, 295(1–4), 225-245.
4. URIBE, M.; LÓPEZ, R.; RAMOS, P. "Validación de métodos analíticos en laboratorios de ensayo de aguas residuales". Revista de divulgación. División Académica de Ciencias Biológicas. 2013, 19(36), 11-18. ISSN-1665-0514.
5. ABU SHAWISH, H. M.; KHEDR, A.; ABED ALMONEM, K.; GABER M. "A comparative study of solid and liquid inner contact benzalkonium chloride ion-selective electrode membranes". Talanta, 2012, 101, 211-219.
6. GRANHOLM, K.; LEWENSTAM, A.; SOKALSKI, T.; IVASKA , A. "Ion-selective electrodes in potentiometric titrations; a new method for processing and evaluating titration data". Analytica Chimica Acta. 2015, 888, 36-43.
7. HU, J.; STEIN, A.; BÜHLMANN, P. "Rational design of all-solid-state ion-selective electrodes and reference electrodes". Trends in Analytical Chemistry. 2016, 76, 102-114.
8. APHA, AWWA, WEF. Standard Methods for the Examination of Water and Wastewater. 21 Edición. Washington, DC, USA, 2005.
9. EURACHEM GUIDE. The Fitness for Purpose of Analytical Methods. A Laboratory Guide to Method Validation and Related Topics. 2da Edición, B Magnusson, U Örnemark Eds, 2014. ISBN 978-91-87461-59-0.
10. OFICINA NACIONAL DE NORMALIZACIÓN. Guía para la validación de métodos de ensayos químicos para alimentos. La Habana: NC TS 368:2 004, 2004.
11. OFICINA NACIONAL DE NORMALIZACIÓN. Guía para la validación de métodos de ensayos químicos para alimentos. La Habana: NC TS 368:2010, 2010.
12. FERNÁNDEZ, M. Desarrollo y validación de métodos analíticos para la determinación de distintas matrices medioambientales de tensioactivos aniónicos derivados de alcoholes: alcoholes sulfatos y etoxisulfatos. Tesis doctoral, Universidad de Granada, España, 2011.
13. KUTNER, M.; NACHTSHEIM, C.; NETER, J.; LI, W. Applied Linear Statistical Models. 5ta Edición. Minesota: The McGraw-Hill, 2005. ISBN 0-07-238688-6
14. DE BEER, J.; NAERT, C.; DECONINCK, E. "The quality coefficient as performance assessment parameter of straight line calibration curves in relationship with the number of calibration points". Accred Qual Assur. 2012, 17(3), 265–274.
15. VAN LOCO, J.; ELSKENS, M.; CROUX, C.; BEERNAERT, H. "Linearity of calibration curves: use and misuse of the correlation coefficient". Accred Qual Assur. 2002, 7(7), 281–285.
16. IUPAC. Harmonised Guidelines for the In–House Validation of Methods of Analysis (technical report). pp. 4-34. Budapest. 2012.
17. CUADROS RODRÍGUEZ, L.; GARCÍA CAMPA, A. M.; JIMÉNEZ LINARES, C.; ROMÁN CEBA, M. "Estimation of Performance Characteristics of an Analytical Method Using the Data Set Of The Calibration Experiment". Analytical Letters. 1993, 26(6), 1243-1258.
18. ROZET, E. et al. "Analysis of recent pharmaceutical regulatory documents on analytical method validation". Journal of Chromatography A. 2007, 1158(1-2), 111-125.
19. MURRAY, E. et al. "A colorimetric method for use within portable test kits for nitrate determination in various water matrices". Analytical Methods. 2017, 9(4), 680-687.
20. WANG, Q. et al. "Methods for the detection and determination of nitrite and nitrate: A review". Talanta. 2017, 165(1), 709-720.
21. LOPEZ MORENO, C.; VIERA PEREZ, I.; URBANO, A. M. "Development and validation of an ionic chromatography method for the determination of nitrate, nitrite and chloride in meat". Food Chemistry. 2016, 194(1), 687-694.
22. SAH, R. N. "Nitrate-nitrogen determinationa critical review". Communications in Soil Science and Plant Analysis. 1994, 25(17-18), 2841-2869.
23. GARFIELD, F. M. Principios de garantía de calidad para laboratorios analíticos. AOAC International. Madrid: Edición Española, 1993.
24. JURADO, J. M. Aplicación de Microsoft Excel a la Química Analítica: validación de métodos analíticos [Fecha de consulta: abril 2008]. [Disponible en: http://personal.us.es/jmjurado/docs/AQAEXCEL.pdf]
25. NORMA CUBANA OBLIGATORIA, NC: 827/2010. Agua Potable-Requisitos Sanitarios. Oficina Nacional de Normalización. La Habana. 2012.
Recibido: 24/04/2017
Aceptado: 4/07/2017
MSc. Surey Ramírez-González, Centro de Estudio de Química Aplicada, Universidad Central de Las Villas, Cuba, sureyrg@uclv.edu.cu

