










 Serviços customizados
Serviços customizados
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
Introducción
Los hongos se distribuyen ampliamente a nivel mundial y se conocen aproximadamente 10 000 especies, de las cuales solo el 10% es comestible, entre estas se encuentran las del género Pleurotus. Dentro de este género se distingue P. ostreatus (Jacq. ex Fr.) P. Kum., especie cosmopolita con un elevado valor nutricional y propiedades terapéuticas, además de una amplia gama de aplicaciones biotecnológicas y ambientales.1
Pleurotus ostreatus representa un recurso valioso por su papel en la degradación de residuos lignocelulósicos, al crecer en una gran variedad de materiales vegetales y producir cuerpos fructíferos que constituyen un alimento de alta calidad nutricional.2 También, tienen propiedades medicinales, debido a que sintetizan metabolitos secundarios con promisoria actividad biológica (anticancerígena, inmunomoduladora, antitumoral, antioxidante y antibacteriana). Dichas propiedades hacen de este hongo uno de los más cultivados en el mundo y de los más atractivos para el mercado. Sin embargo, existen factores que contribuyen a la pérdida de calidad de las setas y disminuyen su valor comercial.3 Uno de ellos es el cambio de coloración que ocurre durante su manipulación en las etapas de recolección, y almacenamiento. Estas reacciones tienen lugar como resultado de la oxidación de sustratos fenólicos en quinonas, y que rinden como producto final la melanina.4
La melanina es un pigmento ubicuo, de color pardo y alto peso molecular, responsable de la coloración de la piel, la protección contra los rayos ultravioleta y las radiaciones ionizantes, participa en la defensa inmunológica, y en otras funciones biológicas. Dicho pigmento se produce por la polimerización de dopaquinonas mediante la tirosinasa (TY) EC 1.14.18.1, enzima que cataliza la monooxigenación de la tirosina.5
La tirosinasa cataliza la hidroxilación de monofenoles a o-difenoles (actividad monofenolasa/cresolasa) y la oxidación de o-difenoles a o-quinonas (actividad difenolasa/catecolasa). En las plantas encontramos otras enzimas similares, las catecol oxidasas (CO), que catalizan solo la oxidación de los o-difenoles.5 Las TY y CO, comúnmente conocidas como polifenoloxidasas (PPOs), pertenecen al grupo de proteínas de cobre tipo 3.3,6 En ambas, el centro bicúprico está rodeado por un paquete de cuatro α hélices y coordinado por seis histidinas.5 Una estructura similar ha sido reportada en la tirosinasa de Streptomyces castaneoglobisporus y en la hemocianina de moluscos y artrópodos.7
El sitio activo de la TY y la CO durante el ciclo catalítico existe en tres estados de oxidación diferentes. La forma met [Cu(II)-Cu(II)] corresponde al estado de reposo de la enzima, donde los iones Cu[II] interactúan con ligandos pequeños como el agua o iones OH-. La forma deoxi [Cu(I)-Cu(I)] es una especie reducida que se une al oxígeno y da lugar a la tercera forma, oxi [Cu(II)O22--Cu(II)]. En este último estado, el oxígeno molecular se une como peróxido en una configuración que desestabiliza y activa el enlace O-O.7
La actividad catecolasa ha sido estudiada en plantas y hongos. Sin embargo, la actividad tirosinasa solo ha sido demostrada para un limitado número de estas enzimas. Se ha propuesto que todas las COs de plantas y hongos poseen actividad monofenolasa, pero no existe consenso sobre esta hipótesis. Por esta razón, aún no está completamente esclarecido el papel fisiológico de la actividad monofenolasa en dichos organismos.8 Recientemente, el grupo Fishman presentó la estructura cristalina de la tirosinasa de Bacillus megaterium con y sin sustratos mono- o difenólicos unidos. Estos autores sugieren que la discriminación funcional entre la tirosinasa y la catecol oxidasa se debe a una Asparagina que junto con un Glutamato, se unen y activan una molécula de agua conservada, hacia la desprotonación de mono-fenoles, que se requiere para la actividad tirosinasa.5
Aunque la función de la tirosinasa en la pigmentación de mamíferos ha sido ampliamente estudiada y bien caracterizada,9 aún no está clara la función y activación in vivo de las polifenol oxidasas.8 El oscurecimiento de frutas, vegetales y hongos mediado por las polifenoloxidasas, como resultado de la formación de melanina después de un daño mecánico, sugiere un papel en la defensa contra insectos y patógenos.4 Estos efectos del oscurecimiento enzimático pueden contrarrestarse con compuestos químicos inhibidores que eviten el contacto del sustrato con el sitio catalítico de la proteína. Entre los inhibidores de tirosinasa se han reportado algunos triterpenoides y flavonoides presentes en plantas y otros organismos. Estos compuestos son capaces de inhibir la síntesis de la melanina.10
Existe limitada información sobre la estructura de la tirosinasa hipotética de P. ostreatus, así como de las mínimas diferencias estructurales y funcionales entre las tirosinasas y catecol oxidasas.11 En este contexto, el uso de métodos computacionales para la predicción de estructuras de proteínas, como la modelación por homología, puede aportar información de utilidad. El objetivo de este trabajo estuvo enfocado a obtener un modelo por homología de la tirosinasa hipotética de Pleurotus ostreatus PC15, para realizar futuros ensayos in silico con sustancias reportadas como inhibidoras del oscurecimiento enzimático, producido después de la cosecha de las setas.
Materiales y métodos
Obtención y análisis de la secuencia de aminoácidos
La secuencia de aminoácidos de la tirosinasa de P. ostreatus, con número de acceso KDQ24083, se obtuvo de las bases de datos sobre secuencias y funciones de proteínas del Centro Nacional de Información Biotecnológica de los Estados Unidos (NCBI), a partir de la secuencia en formato Fasta, se realizó la evaluación del contenido porcentual para los aminoácidos mediante la herramienta GPMAW Lite. El programa permite conocer la composición de residuos de aminoácidos, identificar aminoácidos específicos ácidos o básicos y predecir, además, las modificaciones post-traduccionales.
Búsqueda de plantillas
Para encontrar secuencias plantillas potencialmente similares a la de interés, se utilizó el algoritmo BLAST (Herramienta de Alineamiento Básico Local), a partir del servidor de la base de datos Protein Data Bank (PDB). Los parámetros de alineamiento se muestran en la Tabla 1. Además, para validar los resultados del BLAST, se utilizó el servidor del Swiss model en su opción "buscar plantillas" en la librería. La librería del Swiss model utiliza tanto BLAST como HHblits, como buscadores; para identificar las plantillas y obtener los alineamientos entre la proteína a modelar y su plantilla. El uso combinado de estos dos métodos garantiza el alineamiento correcto, incluso cuando no hay alta identidad de secuencia (homología) entre el modelo y su plantilla. A partir de 89 plantillas, se seleccionó la plantilla 4j3p A con 40.92% de identidad de secuencia.
Predicción de la estructura secundaria
Basada en la secuencia de aminoácidos, la predicción de las estructuras secundarias se realizó mediante el servidor JPRED 4. Este servidor permitió clasificar cada aminoácido según tres posibles estados de estructura secundaria: α-hélice, hoja β u otros. A la predicción de cada residuo se le asignó un índice de fiabilidad, que varía entre 0 y 9, y que se correlaciona con la exactitud de la predicción. Este índice permite identificar las regiones de la proteína que se han predicho con mayor exactitud.12 Posteriormente, se analizaron los dominios de la tirosinasa hipotética a través del servidor Pfam.
Construcción, descripción y evaluación del modelo 3D de la proteína
A partir de la secuencia KDQ24083 de la tirosinasa se construyeron modelos teóricos a través de Swiss Model,13 I-TASSER,14 ITASSER y Phyre2,15 Se utilizaron tres servidores para seleccionar el modelo más preciso según los mejores valores de ajuste. Las tres herramientas construyeron las estructuras terciarias conforme a los parámetros de similitud, identidad y cobertura, los cuales fueron analizados y contrastados con base a los criterios de aceptación.
Se evaluó la calidad de los modelos por medio de Procheck. Se consideraron las restricciones de los ángulos diedros φ y ψ, proporcionadas por el diagrama de Ramachandran según la herramienta Procheck.16 Este diagrama proporciona la distribución de los ángulos de torsión de una estructura proteica y permite pronosticar las regiones favorecidas, permitidas y no permitidas para estos ángulos. Lo cual sirve como indicador de la calidad de los modelos de la estructura tridimensional de una proteína.17
El servidor ProSA, permitió calcular el z-score, el cual indica la calidad general del modelo y la energía en función de la posición de los aminoácidos en la secuencia. La calidad del modelo general se determinó al comparar el puntaje obtenido con el valor correspondiente de proteínas de tamaño similar cristalizadas por rayos X o analizadas en solución por Resonancia Magnética Nuclear (RMN). Por otro lado, el análisis energético de las secuencias facilitó la detección de regiones con energía positiva. Este último análisis se realizó por bloques de 10 y 40 aminoácidos para calcular la energía promedio de cada posición en la secuencia. Se utilizó Verify3D, para calcular el perfil 3D de la proteína y considerar la compatibilidad del modelo atómico (3D) con su propia secuencia de aminoácidos (1D). El modelo debe tener más del 80% de los residuos de aminoácidos con puntaje ≥ 0.2 en el perfil 3D/1D.
Resultados y discusión
Obtención y análisis de la secuencia de aminoácidos de la proteína tirosinasa
Mediante la búsqueda realizada en el NCBI se obtuvo la secuencia de aminoácidos en formato FASTA de la enzima tirosinasa hipotética, constituida por 337 residuos de aminoácidos. Por su parte, GPMAW Lite identificó cinco modificaciones postraduccionales, correspondientes a N glicosilaciones (figura 1).

Fig. 1 Composición aminoacídica y de los sitios de N-glicosilación de la enzima mediante colores. Fuente: GPMAW Lite.
La secuencia consenso más representativa para N-glicosilación es N-X-S/T, donde X se corresponde con los aminoácidos Valina, Leucina, Metionina o Triptófano.18 Los sitios de glicosilación se encuentran ubicados generalmente en zonas de giros y lazos, lo cual pudiera estar relacionado con el acceso para la transferencia del núcleo por parte del complejo Oligosacaril Transferasa (OST). Este complejo proteico es el encargado de transferir el núcleo desde el precursor lipídico a un residuo de asparagina de la secuencia consenso de la proteína.18
El plegamiento adecuado de la tirosinasa en el retículo endoplásmico parece ser crucial para su posterior transporte al aparato de Golgi. Cuando ocurren errores en el plegamiento de la proteína en el retículo endoplásmico, la misma no se transporta al aparato de Golgi y puede ser degradada por el proteosoma. En el complejo trans-Golgi se procesan los glicanos y se adquieren los iones cobre necesarios para la activación de la enzima, aunque este evento parece ser independiente del plegamiento de proteínas. Por otro lado, en tirosinasas de mamíferos se ha demostrado que el plegamiento correcto de la proteína es necesario para el funcionamiento de la tirosinasa.19
La masa aproximada de la enzima es 37, 520,11 Da y en su composición predominan los aminoácidos neutros (69,76 %), seguidos por los aromáticos (10,98 %), los básicos (9,79 %) y los ácidos (9,47%). Asimismo, la glicina resultó el aminoácido más frecuente (8,61 %) y la cisteína el menor frecuencia (1,78%).
Selección y análisis de la plantilla
Según la exploración en PDB, hasta julio del 2019, se habían registrado 35 estructuras tridimensionales homólogas. Dichas estructuras, resueltas por difracción de rayos X, se obtuvieron de 14 especies. Pertenecientes a tirosinasas de Streptomyces castaneglobisporus, Bacillus megaterium, Burkholderia thailadensis y Agaricus bisporus var. bisporus; polifenol oxidasas de Vitis vinifera, Juglans regia, Solanum lycopersicum y Malus domestica; hemocianina de Rapana venosa, Megathura crenulata y Enteroctopus dofleini; auresidina sintetasa de Corepsis grandiflora y catecol oxidasa de Ipomea batatas y Aspergillus oryzae. Por otro lado, no se encontró ninguna estructura tridimensional resuelta perteneciente a Pleurotus spp.
Los mayores porcentajes de identidad (36,57 %), cobertura (96 %) y el valor de error (E=8x10-64) más bajo pertenecen a las estructuras 4j3p y 6GSG de Aspergillus oryzae (tabla 2). El valor de E representa el número de alineamientos fortuitos que puede esperarse entre la secuencia en estudio y cualquier otra de las existentes en una base de datos. Por lo tanto, entre más cercano a 0 sea el valor de E, más significativo es el alineamiento entre la secuencia que se analiza y la relacionada.20
Tabla 2 Resultados obtenidos del BLAST para la búsqueda de homología de la tirosinasa de Pleurotus ostreatus utilizando la matriz de sustitución BLOSUM 62.
| Código PDB | Organismo | Proteína | Resolución (Å) | % Cobertura | % Identidad | Error |
| 4j3p A | Aspergillus oryzae | catecol oxidasa | 2,5 | 96 | 36, 57 | 8e-64 |
| 6gsg A | Aspergillus oryzae | catecol oxidasa | 2,19 | 96 | 36, 57 | 8e-64 |
| 4j3q A | Aspergillus oryzae | catecol oxidasa | 2,9 | 91 | 36, 69 | 1e-63 |
Una nueva búsqueda en BLAST, a partir de la matriz de sustitución BLOSUM 45, resultó en la obtención de moldes similares, aunque con orden diferente en las estructuras 4j3p A y 4j3q A. En la matriz BLOSUM los valores se basan en la frecuencia de sustitución en bloques de alineamientos locales para proteínas relacionadas evolutivamente, de esta manera se enfoca en regiones conservadas. BLOSUM 62 y BLOSUM 45 se refieren a alineamientos con un máximo de 62% o 45% de identidad entre las secuencias comparadas.21
Cabe destacar que para la matriz de sustitución PAM 200 el resultado difirió del obtenido en las matrices BLOSUM 62 y BLOSUM 45. Estos resultados resultan lógicos, debido a las diferencias existentes entre las matrices PAM y BLOSUM. Las primeras se basan en un modelo explícito de la evolución y mientras más grande sea el valor de la matriz, la distancia evolutiva es mayor. En la actualidad, la matriz BLOSUM 62 es la más utilizada.20
Para la búsqueda con el Swiss model, según BLAST, los mayores valores de cobertura (93%) e identidad (41,8%) correspondieron a la estructura con código y resolución de 2.50 Å. A continuación, se registró la estructura con código 4j3q A y resolución de 2.90 Å, cuya cobertura alcanzó 90 % y la identidad, 40,92 %. La estructura 4j3p A se seleccionó como plantilla para construir el modelo de la tirosinasa hipotética de Pleurotus ostreatus, basado en el porcentaje de similitud entre ambas. El análisis de las similitudes y diferencias en aminoácidos individuales busca inferir relaciones estructurales, funcionales y evolutivas entre las secuencias en estudio. La explicación más simple para la elevada similitud, es que las dos secuencias surgieron de un antepasado común.20 Por otro lado, la búsqueda con HHblist arrojó valores inferiores de identidad y similitud para ambas estructuras con respecto a los de BLAST.
Alineamiento de plantilla y secuencia de tirosinas a hipotética
En la figura 2 se muestra el alineamiento múltiple entre la plantilla y la tirosinasa hipotética. El alineamiento de secuencias muestra un patrón discontinuo con brechas. Se conservan los residuos de Histidina (H) H 80, H 88, H 97, H 242, H 246 y H 269, los cuales probablemente se unen de manera coordinada a dos átomos de cobre CuA y CuB y forman el sitio activo de la tirosinasa. En esta también se encuentran otros residuos de aminoácidos funcionales como parte de motivos conservados en el dominio central como la Prolina, Glicina y Fenilalanina. Se identificaron dos residuos de Fenilalanina próximos a cada átomo de CuA, lo que pudiera relacionarse con la pérdida de la actividad monofenol oxidasa o monofenolasa de la catecol oxidasa.

Análisis de la estructura secundaria
Los posibles plegamientos de la estructura secundaria indicaron la presencia, en mayor proporción, de estructuras en forma de siete lazos (78,91 %), cuatro hélices α (18,69 %) y cuatro de láminas β (2,4 %). Las hélices α mostraron porcentajes de confiabilidad mayores que siete (figura 3). Para el resto de la estructura secundaria, el “nivel de confiabilidad” se mantuvo preferentemente por encima de cinco. La estructura obtenida tiene un nivel intermedio de confiabilidad. El predominio de estructuras en forma de lazos, sugiere una estructura secundaria poco definida.22

Identificación de dominios
El dominio tirosinasa, catalogado como el dominio central de las proteínas PPO fue identificado a través de la herramienta Pfam. Este abarcó los aminoácidos comprendidos entre las posiciones 70 y 282, e incluye el centro activo de la tirosinasa.
Construcción, descripción y evaluación de los modelos
La construcción del modelo con la herramienta Swiss-model permitió obtener una proteína con identidad de secuencia de 41,8 %, similitud de 0,41 y cobertura de 0,93. El QMEAN fue de -4,59 y el valor normalizado estuvo dentro de los límites de z-score menor que 1, lo cual indica la confiabilidad del modelo.13
A través de ITASSER se obtuvieron tres modelos potenciales de la estructura terciaria de la tirosinasa. La identidad de la secuencia con la plantilla fue de 40 %, la cobertura de 0,99, TM de 0,83±0,13 y el puntaje c-score estuvo en el rango entre -5 y 2. Para este puntaje los valores más positivos indican un modelo con alta confianza,14 aunque el modelo 1 resultó el mejor con un valor de c-score de 0,85.
Con el objetivo de escoger el modelo más estable estereoquímicamente se obtuvieron sus diagramas de Ramachandran con la herramienta Procheck. En este caso, se seleccionó el modelo 1por presentar mayor porcentaje de residuos ubicados en regiones favorables, 84,3 %. Además, con el servidor Phyre2 se detectó 37 % de identidad entre la secuencia y la plantilla, la cobertura fue 98 % y 100 % de confianza.
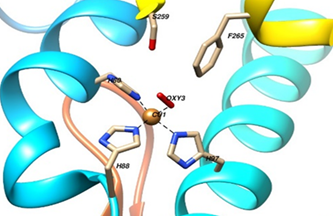
Para los tres servidores, los modelos de la tirosinasa hipotética de P. ostreatus PC15 son similares a sus proteínas homólogas en hongos (catecol oxidasas). Cada modelo es un homodímero, con dos sitios idénticos de unión a metales. Estos centros están compuestos por un átomo de Cu en su forma natural y con algunos residuos conservados en el sitio activo. Dicho sitio está formado por tres histidinas y una molécula de agua. Esta región se asemeja a una esfera hidrofóbica que protege al sitio activo y se pliega en un bulbo de cuatro α hélices, con los sitios de unión al cobre, excepto la histidina (H) H88 (figura 4). El ion CuA es coordinado por H 80, H 88, H 97; mientras el ion CuB por H 242, H 246 y H 269.

Fig. 4 Sitio activo de la tirosinasa hipotética de Pleurotus ostreatus PC15 contenido en cuatro α hélices. Fuente: UCSF Quimera. Versión: 1. 13. 1.
El sitio activo se encuentra protegido por dos residuos de aminoácidos, Fenilalanina F 265 y Serina S 259. El residuo voluminoso de Fenilalanina se ubica sobre el centro activo, específicamente sobre el CuA. Este residuo funciona como “puerta” al bloquear el acceso al sustrato (figura 5). El bloqueo del sitio A por la Fenilalanina, se relaciona con la pérdida de actividad monofenolasa de las catecol oxidasa. La función del residuo de S 259 aún no está clara, pero pudiera relacionarse con la activación del sustrato. Dicho residuo de F 265 pudiera ser una explicación a las diferencias entre catecol oxidasas y tirosinasas.6,23
Investigaciones realizadas con la tirosinasa de Bacillus megaterium y otros estudios del mecanismo catalítico de las polifenoloxidasas indicaron que los sustratos fenólicos, tanto monofenoles como difenoles, se unen de manera idéntica hacia el CuA en el sitio activo. Sin embargo, solo los monofenoles rotan durante la reacción, esto constituye una diferencia entre las enzimas con actividad mono-difenolosa y las enzimas con actividad estrictamente difenolasa. En la catecol oxidasa de Ipomea batatas, fue descubierta una restricción estructural de la actividad monofenolasa, un residuo voluminoso de F 261, que no está presente en tirosinasas. En el caso de las enzimas tirosinasas, estaría sustituido por uno menos voluminoso correspondiente a Ala o Val. La presencia de la F 261 impide la rotación del sustrato durante la reacción, necesaria para la actividad monofenolasa, ocurriendo una reorientación del sustrato hacia el CuB.24
Una explicación alternativa para las diferencias catalíticas entre las tirosinasas y catecol oxidasas podría ser la presencia de un enlace tioéter en las catecol oxidasa, el cual limita la flexibilidad de la Histidina e impide la rotación del sustrato durante la reacción.9 Probablemente, la reacción del sustrato L-Dopa no implica ataque electrofílico sino la oxidación de dos grupos hidroxilo y su rotación no es necesaria. Por otro lado, se ha demostrado que la discriminación funcional entre las tirosinasas y las catecol oxidasas se debe exclusivamente a la presencia de una Asparagina que, junto con el Glutamato, se une a una molécula de agua conservada.5 Como resultado, la molécula de agua conservada se activa y se une al protón liberado por sustratos fenólicos después de la coordinación con el sitio activo.5
Evaluación del modelo
Los parámetros de calidad, estimados según la herramienta Procheck, indicaron que las estructuras 3D calculadas resultaron adecuadas. Dichos parámetros fueron similares a los de la estructura de partida 4j3p.1.A (tabla 3). Los valores fueron cercanos al 90 % de residuos de aminoácidos para las regiones más favorecidas en el diagrama de Ramachandran, lo cual valida los tres modelos propuestos. Los diagramas de Ramachandran señalan a Tyr Swiss como mejor modelo con 85,6% de residuos en regiones más favorecidas (figura 6A).25 En el resto de los modelos los porcentajes de residuos fueron menores, Tyr Tass (84,4%) y Tyr Phy2 (84,3%).
Tabla 3 Resumen de los gráficos de ramachandran. Porcentajes de residuos ubicados
| Criterio | Parámetro | 4j3p.1.A | Tyr Swiss | Tyr Tass | Tyr Phy 2 |
| Procheck | Más favorecidas Permitidas adicionales Permitidas generosamente No permitidas | 89,0% 10,6% 0,0% 0,3% | 85,6% 11,2% 1,1% 2,1% | 84,3% 11,0% 2,2% 2,5% | 84,4% 10,6% 3,5% 1,4% |

Fig. 6 A. Tyr Swiss, B: Tyr Tass, y C: Tyr Phy2. Diagramas de Ramachandran del homodímero del modelo de la proteína tirosinasa de Pleurotus ostreatus. Fuente: Procheck.
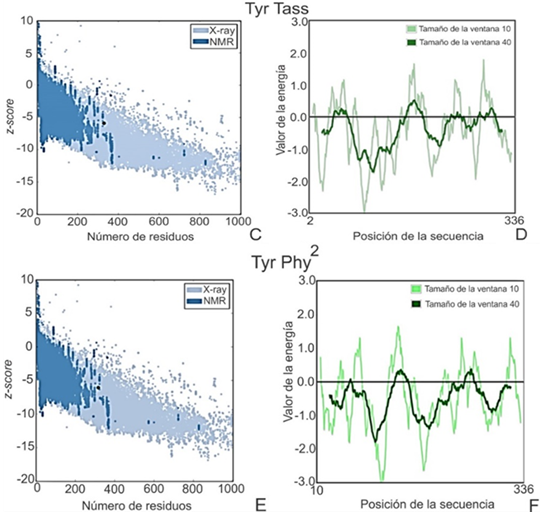
La determinación de z-score a través del servidor ProSA 6 indicó que los modelos de tirosinasa de P. ostreatus poseen puntuaciones globales de -7,15, -6,06 y -5,91, respectivamente (figura 7 A, C y E). Según estos valores los modelos se ubican en una zona de aceptación. Mediante ProSA 26 también se evaluó la calidad local de la estructura según la longitud de la secuencia; el análisis del perfil energético con bloques de 10 resultó el más efectivo puesto que separó mejor las “regiones problemáticas”. Los perfiles energéticos de los tres modelos mostraron zonas con energía positiva (figura 7 B, D y F). Este patrón pudiera relacionarse con la presencia de un lazo más extenso en esa zona de la estructura. Un diseño con estas características aseguraría mayor movilidad a la región e incrementaría su energía. No obstante, el modelo Tyr Swiss resultό más estable energéticamente (figuras 7A y B).

Perfil tridimensional del modelo por Verify3D
Luego de seleccionado el Modelo Tyr Swiss como el más estable, se procedió a realizar los análisis de la distribución del perfil 3D por residuo. Como promedio, el 80,73 % de los residuos alcanzó puntaje mayor o igual que 0,2; en el perfil 3D/1D, lo cual indica que el entorno del perfil tridimensional del modelo es bueno. Consecuentemente, el modelo puede considerarse compatible con su secuencia primaria de aminoácidos.
Conclusiones
Se obtuvieron tres modelos tridimensionales para la tirosinasa hipotética de Pleurotus ostreatus PC15. Los servidores de modelado por homología: Swiss model, ITASSER Y Phyre2 resultaron herramientas computacionales útiles para el diseño estructural de la tirosinasa hipotética P. ostreatus. El modelo Tyr Swiss, a través de la evaluación realizada con los servidores Procheck, ProSA y Verify3D, puede ser considerado como un modelo válido con potenciales aplicaciones en el campo de investigaciones biotecnológicas como referencia estructural, para selección de inhibidores de la proteína tirosinasa de utilidad industrial

