










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Ciencias Informáticas
versión On-line ISSN 2227-1899
Rev cuba cienc informat vol.7 no.3 La Habana jul.-sep. 2013
ARTÍCULO ORIGINAL
Modelación y manejo de bases de datos para el almacenamiento de la información sobre ortología genética
Modeling and management of databases for storing information about genetic orthology
Tonysé de la Rosa Martín1, Deborah Galpert Cañizares2, Mario Pupo Meriño2
1 Universidad de las Ciencias Informáticas, Carretera a San Antonio de los Baños, km 2 ½, Torrens, Boyeros, La Habana, Cuba. C.P.: 19370. E-mail. ajardines@uci.cu
2 Universidad Central Marta Abreu de las Villas. Carretera a Camajuaní km 3½. Santa Clara, Villa Clara. Cuba. E-mail. deborah@uclv.edu.cu
3 Departamento de Informatización De Entidades, CEIGE, UCI, La Habana, Cuba. E-mail. mpupo@uclv.edu.cu
RESUMEN
La presente investigación tiene como precedente la necesidad de crear bases de datos locales para el almacenamiento de información referente a la ortología genética y secuencias genómicas de especies para el posterior estudio de éstas por los investigadores del Centro de Estudios de Informática (CEI) de la Universidad Central Marta Abreu de las Villas (UCLV). En la investigación se obtiene una aplicación informática desarrollada a partir de tecnologías libres que integra los procesos de validación e incorporación de información a partir de ficheros XML (Lenguaje de marcado extendido, por sus siglas en inglés) de secuencias genómicas y de información ortológica, así como la creación de distintos tipos de ficheros utilizados por otras aplicaciones dentro del área de la Bioinformática. Se incluye el estudio de las tecnologías y herramientas necesarias para el diseño e implementación de las bases de datos creadas con este fin, así como de la aplicación informática para el manejo de la información contenida en estas bases de datos. Se presenta la prueba del sistema en cuanto a su correcto funcionamiento, evidenciando que la utilización del mismo contribuirá a la disminución de las dificultades del uso de aplicaciones de manejo de ortología en internet por el tiempo de procesamiento y descarga de datos de gran volumen.
Palabras clave: Ortología genética, Bases de datos biológicas, Ficheros XML, Ficheros orthoXML, Ficheros seqXML.
ABSTRACT
This previous research arise under the need to create their own databases for storing information regarding the orthology genetic and genomic sequences of species for further study of researchers at the Center of Informatics Studies (CIS) of the "Martha Abreu" Central University of Las Villas (UCLV). This investigation presents the results of the design and implementation of two databases from free technologies that integrate validation processes and incorporation of information from XML files genomic sequences and ortological information. To achieve the objectives outlined above study of technologies and tools for the design and implementation of databases created for this purpose, as well as the application for the management of the information contained in these databases. Evidence is presented in terms of the system function properly, showing that the use of it will help to reduce the difficulties of using orthology handling applications online by processing time and download large data volume.
Key words: Genetic Orthology, Biological Databases, XML Files, orthoXML Files, seqXML Files .
INTRODUCCIÓN
El mundo se mueve desde el último medio siglo inmerso en una época que tradicionalmente ha sido llamada la era de la Ciencia, y la sociedad de tal era no puede comprenderse sin destacar una de esas múltiples disciplinas científicas, que sin duda, es y será un referente para la investigación futura: la Bioinformática.
La Bioinformática, llamada también Biología Molecular Computacional, corresponde como tal a una disciplina científica que utiliza tecnología de la información para organizar, analizar y distribuir información de Biomoléculas con la finalidad de responder preguntas complejas (Altschul, Boguski, Gish, & Wootton, 1994). Representando así una respuesta alternativa y eficaz a los nuevos problemas en materia de salud que surgen en la sociedad actual.
Nos encontramos en el período post-genoma humano conocido también como período de las ómicas: la Genómica, la Proteómica, la Metabolómica y la Citómica, que conllevan una gran cantidad de datos a analizar. La secuenciación masiva de genomas, la determinación de estructuras de proteínas, el seguimiento, la expresión de miles de genes de manera simultánea, la determinación de interacciones entre proteínas y el estudio sistemático de las modificaciones post-traduccionales de proteínas separadas en genes 2-D y caracterizadas por espectrometría de masas llevan a generar insospechados datos, de los que se conocen propiedades globales, pero no en detalle (Martínez, 2010).
De ahí que el protagonismo de la Bioinformática está dado por dos razones principales: la primera es que la enorme cantidad de datos de origen biológico sólo puede ser analizada utilizando computadoras; la segunda es que los datos no están tan comprensibles y las informaciones sólo pueden salir a la luz utilizando sofisticados algoritmos computacionales. Por lo tanto el almacenamiento depurado, control y mantenimiento de estos datos con fines específicos como: el diseño de nuevos fármacos, comparación entre especies por su semejanza génica, entre otras es muy importante para los científicos e investigadores que trabajan en esta disciplina. En este sentido, en el país se han creado un grupo de instituciones dedicadas permanentemente al desarrollo de la Bioinformática y que han adquirido cierta madurez y experiencia en este campo.
Como centro de gran valía y aporte científico en esta rama se encuentra el Centro de Estudios de Informática (CEI) de la Universidad Central Marta Abreu de las Villas, el cual cuenta con un Laboratorio de Bioinformática en el que se realizan diversos estudios en materia de: Inteligencia Artificial aplicada a la Bioinformática, elaboración de árboles filogenéticos y estudios sobre Homología biológica.
La homología biológica no es más que: la relación que existe entre dos partes orgánicas diferentes cuando sus determinantes genéticos tienen el mismo origen evolutivo.
Los estudios han definido dos formas de homología:
• homología primaria es el que implica un investigador, que afirma una creencia de que dos personajes comparten un linaje.
• homología secundaria está implicada por el análisis de parsimonia, conocido también como método de ponderación y medida, donde se toma un carácter que sólo se produce una vez en un árbol para ser homólogo (Pinna, 1991) .
Al igual que con las estructuras anatómicas, la homología entre secuencias de proteínas o de ADN se define en términos de ascendencia compartida. Dos segmentos de ADN pueden haber compartido ancestros ya sea por una especiación (ortólogos) o un evento de duplicación (parálogos) (Koonin, 2005). En la figura 1 se muestra una imagen en la que se ejemplifica cómo a partir del proceso de especiación, el gen A es copiado tanto en el genoma de la especie 1 como en el genoma de la especie 2, convirtiéndose de esta manera en genes ortólogos. En la especie 1 a través de un proceso de duplicación del gen A se obtienen dos genes, un nuevo gen B y el mismo A, convirtiéndose de esta forma el gen B en entidad de tipo paráloga respecto a el nuevo gen A y este mismo gen B se convierte en entidad de tipo homóloga respecto al gen A de la especie 2.
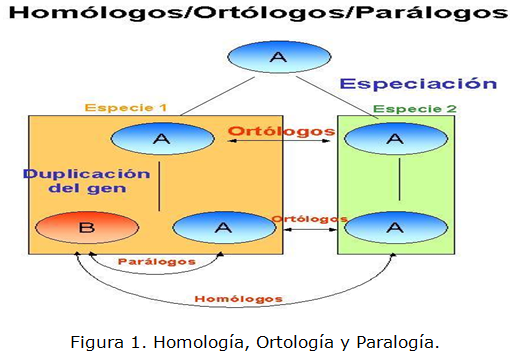
El estudio de estos grupos de genes es sumamente importante para el descubrimiento de funciones semejantes en diferentes especies y así la comprensión de las mismas, con el fin de obtener patrones evolutivos tan necesarios en la predicción y prevención de enfermedades patológicas y genéticas.
El estudio de los grupos de ortología genera una gran cantidad de datos que deben ser almacenados para posteriores estudios, por lo que el almacenamiento de los mismos, de manera segura es muy importante para que puedan ser consultados y utilizados por otros investigadores afines a la rama de la genómica tanto del CEI como de la Comunidad Científica del país.
Actualmente, existe un gran número de científicos y especialistas que realizan estudios sobre la creación de algoritmos y técnicas para obtener grupos de ortologías entre especies, para ser almacenados en grandes bancos de datos. Estas bases de datos son necesarias para comprobar el comportamiento de nuevos algoritmos.
Es importante mencionar que las aplicaciones más avanzadas son comerciales o de propiedad de compañías privadas. La obtención de información de estas bases de datos requiere del uso intensivo de internet de alta velocidad por el volumen de información que manejan. Existe una gran variedad de formatos de los datos tanto en XML como en otras representaciones propias. Algunas aplicaciones disponibles en Internet se han suscrito al proyecto SeqXML-OrthoXML (Schmitt, 2011) como vía de estandarización de la información de ortología.
En algunas investigaciones no es conveniente divulgar la estructura de información, por lo que muchas empresas o grupos de investigación prefieren usar localmente estas bases de datos. Es por esto que investigadores del Centro de Estudios Informáticos (CEI) de la UCLV identificaron la necesidad de contar con una base de datos propia para el almacenamiento de la información de grupos de ortología entre especies y de las secuencias relativas a estas. También el poseer un desarrollo propio en el almacenamiento de los datos tiene la ventaja de poder incorporar nuevas ideas y algoritmos que se deriven de los distintos procesos investigativos.
Materiales y métodos
En el mundo existen muchas bases de datos dedicadas a la recopilación, mejoramiento y análisis de la ortología genética como son: por ejemplo, en eucariotas OrthoMCL (Li, Stoeckert, & Roos, 2012), en mamíferos OrthoMaM (Biomed, 2012) , en plantas OrthologID (Oxford, 2011) y GreenPhylDB (Oxford, 2012) . Otras referenciadas son:
• Ensembl Compara * (Instituto de Bioinformática Europeo).
• InParanoid *(Centro de Bioinformática de Estocolmo).
• MBGD Microbial Genome Database.
• MGD Mouse Genome Database.
• OMA* (Proyecto Matrices de Ortología, Zurich, Suiza).
• OrthoInspector.
• OrthoMCL.
• Panther.
• PHOG.
• PhyloFacts.
• PhylomeDB.
• ProGMap.
Roundup *(Universidad de Harvard) (Stockholm Bioinformatics Center, 2008).
Sucede que muy pocas de ellas brindan la información almacenada y casi ninguna el diseño o la estructura de estas. De las anteriores solo las marcadas con * brindan su información en ficheros de tipo XML (los cuales no poseen un manejo simple para los especialistas). Estos ficheros XML conocidos como orthoXML contienen las relaciones entre grupos de genes ortólogos de dos o más especies especificadas (Stockholm Bioinformatics Center, 2011) y los seqXML (Stockholm Bioinformatics Center, 2008) contienen las secuencias de ADN de los genes de una especie determinada.
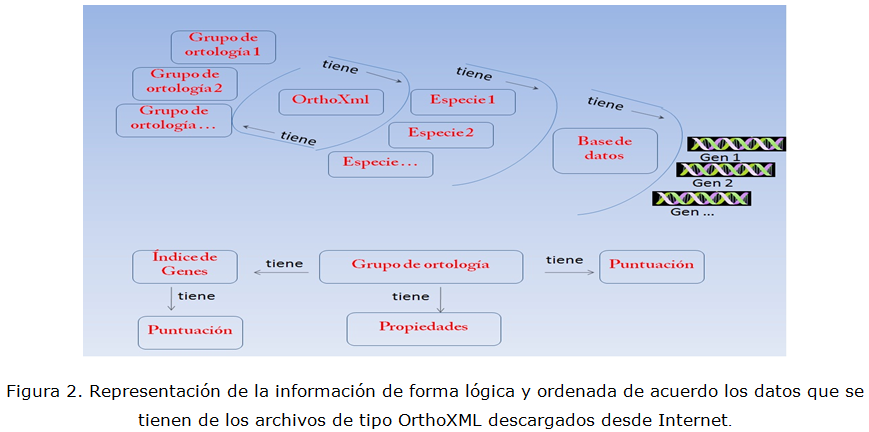
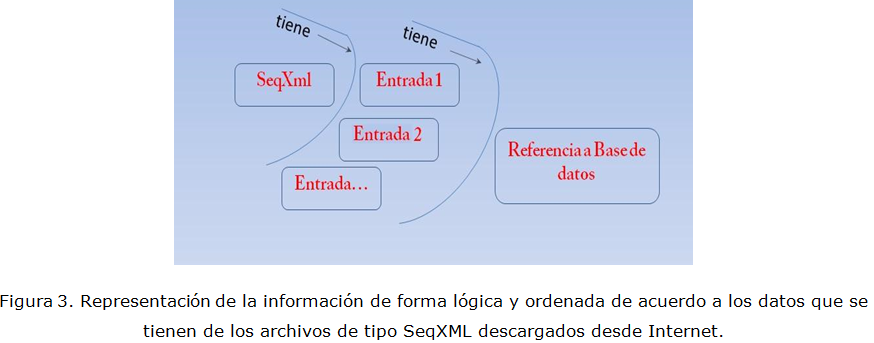
Para realizar este trabajo se ha filtrado la información de los ficheros OrthoXML utilizando el Grafico Escalable de Vectores (SVG) obtenido del proyecto OMA (Ortologic Matrix) (Stockholm Bioinformatic Center, 2011). Por otra parte, la estructura de los ficheros SeqXML es bastante sencilla aunque tiene algunos elementos que son omitidos del diseño como las notas suplementarias y propiedades especiales que son agregadas por los algoritmos, por lo demás su diseño se realizó en conformidad con los criterios de conformación de los grandes proyectos antes mencionados. El ordenamiento lógico y estructurado que se ha realizado de la información almacenada en los ficheros orthoXML y SeqXML se muestran en la figura 2 y figura 3 respectivamente.
En la figura 2 se observa como el fichero OrthoXML posee las especies y cada una de estas posee a su vez una base de datos donde se encuentran los identificadores de los genes que conforman su genoma. También los OrthoXML poseen los grupos de ortología, los cuales poseen los identificadores de los genes ortólogos, la puntuación que los algoritmos asignan a cada grupo y las propiedades del grupo de ortología. Los índices de los genes ortólogos poseen la puntuación de conformación del algoritmo.
En la Figura 3 se observa como el fichero SeqXML posee las entradas y cada una de estas posee a su vez una referencia a la base de datos de donde se extrajeron los datos para la conformación de las secuencias.
Para el diseño e implementación de las bases de datos solo se utilizaron tecnologías y herramientas libres como:
• El Sistema Gestor de Bases de Datos PostgreSQL establecido tanto en el Polo Científico Cubano como en la normativa del estado cubano para el logro de la independencia tecnológica. La versión que se usa del mismo es la 8.4.
• El Visual Paradigm para UML (Lenguaje de Modelado Unificado), herramienta UML profesional que soporta el ciclo de vida completo del desarrollo de software: análisis y diseño orientados a objetos, construcción, pruebas y despliegue. El software de modelado UML ayuda a una más rápida construcción de aplicaciones de calidad, mejores y a un menor coste. Permite dibujar todos los tipos de diagramas de clases, código inverso, generar código desde diagramas y generar documentación, además de crear los artefactos necesarios para el modelado y creación de bases de datos (Visual Paradigm, 2009) .
• La tecnología Java 2 Enterprise Edition (J2EE) que proporciona una completa y potente plataforma orientada al desarrollo de aplicaciones J2EE como plataforma de desarrollo aporta numerosas bibliotecas y ambientes de trabajo, la mayoría de las cuales son gratuitas y de código abierto. Además esta tecnología es multiplataforma (Johnson, 2003).
• El lenguaje Java que ofrece todas las ventajas de un lenguaje potente y robusto, pues fue diseñado para crear software altamente fiable. Es un lenguaje de programación basado en clases y orientado a objetos. Sus características de memoria liberan a los programadores de responsabilidades y errores. Java es compilado en un código intermedio más abstracto que el código de máquina que es ejecutado por la máquina virtual de Java (J2EE) (Arnold, Gosling, & Holmes, 2005).
• UML (Unified Modeling Language), lenguaje que permite modelar, construir y documentar los elementos que forman un sistema de software orientado a objetos. Se ha convertido en el estándar de facto de la industria, debido a que ha sido impulsado por los autores de los tres métodos más usados de orientación a objetos: Grady Booch, Ivar Jacobson y Jim Rumbaugh (Rumbaugh, Jacobson, & Booch., 2004) .
• NetBeans IDE (Entorno integrado de desarrollo, por su siglas en inglés), herramienta para que los programadores puedan escribir, compilar, depurar y ejecutar programas. Está escrito en Java. Existe además un número importante de módulos para extender el NetBeans IDE. Este IDE es un producto libre sin restricciones de uso y es de código abierto y gratuito para uso tanto comercial como no comercial. El código fuente está disponible para su reutilización de acuerdo con la Licencia GPL (Licencia Pública General, por sus siglas en inglés) (Netbeans, 2009).
Para el manejo de las bases de datos OrthoXML y SeqXML se desarrolló una aplicación informática para la inserción y manejo de información proveniente de ficheros de secuencia (seqXML) y de ortología genética (orthoXML) llamada Gregor.
La aplicación visual de este sistema Gregor tiene concebida toda la ejecución de las funcionalidades a partir de una arquitectura cliente-servidor, específicamente en la variante de tres capas, lo que tiene como objetivo compartir información invirtiendo la menor cantidad de recursos. Debido a esto la información que maneja el sistema informático implementado se almacena centralmente en las dos bases de datos anteriormente mencionadas, sin duplicación de información en ninguna de ellas. Los usuarios solo necesitan de una computadora y de la red para utilizar el sistema. El sistema puede ser utilizado por múltiples usuarios a la vez. Las pruebas realizadas al mismo demuestran que pueden estar conectados al menos 40 usuarios a la vez sin que se afecte la integridad y la capacidad de respuesta del sistema.
En el diseño e implementación de las bases de datos del sistema se realizó un análisis a partir del Gráfico Escalable de Vectores (SVG), con el objetivo de determinar cuál sería la mejor estructura para la base de datos OrthoXML; obteniéndose un diseño de menor complejidad estructural que el de las bases de datos revisadas en internet, pues de las más de 21 tablas, con ciclos incluidas en alguna de ellas (ejemplo las del proyecto OMA) y la falta de completitud en campos importantes como es el id del grupo de ortología del proyecto RoundUP, el diseño de la base de datos orthoXML quedó solamente en 13 tablas que recogen la información relevante y necesaria para el desarrollo de esta investigación.
El diseño e implementación de la base de datos de SeqXML por su escasa complejidad estructural y estandarización a nivel mundial se hizo muy semejante a los proyectos antes mencionados incluyendo en estos el Proyecto Inparanoid y el ENSEMBL Compara.
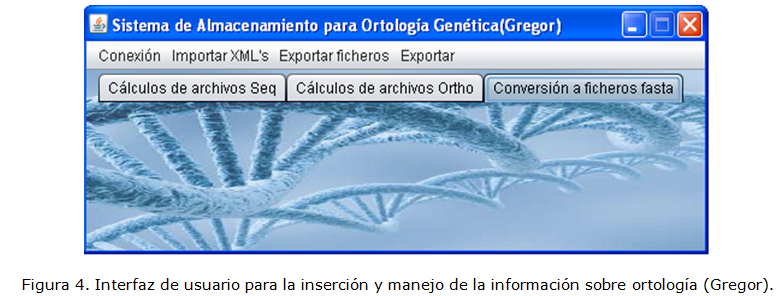
La información que se maneja en la aplicación es pública, pero se mantiene en un ambiente local con la seguridad correspondiente. La interfaz de usuario se muestra en la Figura 4.
El usuario puede agregar los ficheros de las especies de tipo seqXML y orthoXML a las bases de datos a través de la opción Importar XML's. En la opción Exportar ficheros el usuario puede obtener ficheros tipo FASTA y RoundUP a partir de la información contenida en las bases de datos y en la Opción Exportar obtener ficheros de más complejidad como son los de tipo ARFF y SPSS. Las bases de datos SeqXML y OrthoXML han sido pobladas con la información contenida en 1109 ficheros de tipo seqXML y 15 de tipo orthoXML de 28 especies.
Algunas ventajas logradas con el diseño e implementación de las bases de datos son:
• Independencia de Internet cuando se usa localmente en una institución que así lo decida, aunque puede hacerse una herramienta pública que se acceda desde la web.
• Conexión a una base de datos segura, solo accedida por usuarios específicos a través del sistema Gregor. Esto posibilita que se mantenga la confiabilidad de la información y se mantenga disponible siempre que se necesite.
Constituyen aportes a considerar:
• A través del sistema informático implementado para el manejo de las bases de datos creadas se entrega la posibilidad a los especialistas de prescindir de las bases de datos públicas en internet, dado los inconvenientes que conlleva conectarse a estas desde Cuba.
• A partir de las bases de datos se crean un conjunto de funcionalidades dedicadas a facilitar el trabajo de los especialistas en la rama de la ortología genética, a la hora de usar otras aplicaciones como son WEKA, SPSS, etc.
Los especialistas cuentan con una base de datos, sin tener la necesidad de divulgar la información de investigaciones propias y la creación de nuevas ideas a partir de las estructuras almacenadas.
CONCLUSIONES
En la investigación se creó una base de datos (OrthoXML) la cual mejora estructuralmente la información obtenida a partir de los ficheros XML de distintos proyectos dedicados a la conformación y almacenamiento de grupos de ortología en internet.
La base de datos (SeqXML) implementada permite la creación de ficheros de tipo .fasta a partir de los ficheros XML de secuencia de los proyectos dedicados a la conformación de grupos de ortología.
Las funcionalidades implementadas en la aplicación visual (Gregor) permiten la conformación de ficheros de tipo FASTA, ARFF y SPSS, sirviendo de soporte a otras aplicaciones de la rama de la Bioinformática.
El trabajo continuará con la incorporación de nuevos datos procedentes de otras bases en Internet y en la utilización de los mismos en las investigaciones.
AGRADECIMIENTOS
• Al grupo Biosoft del Centro DATEC de la Universidad de la Ciencias Informáticas.
• Al grupo de Bioinformática de la Universidad Central de las Villas Marta Abreu.
REFERENCIAS BIBLIOGRÁFICAS
Altschul, S., Boguski, M., Gish, W., & Wootton, J. (1994). Issues in searching molecular Sequence databases. Nat Genet. 6: P. 119-29 Revisado Noviembre 2011.
Arnold, K., Gosling, J., & Holmes, D.Java (TM) Programming Language. The. Addison-Wesley Professional. 2005.
Biomed, C.BiomedCentral. Disponible en: [http://www.biomedcentral.com/] 1471-2148/7/241: BMC Evol. Biol. 7: p. 241. 2012.
Johnson, R. Expert one-on-one J2EE Design and Development. Wrox. 2003.
Koonin, E. "Ortólogos, parálogos y la genómica evolutiva. Annu. Rev. Genet 39. 2005.
Li, L., Christian J., S., & David, S. R. OrthoMCL-DB. 2012. Disponible en: [http://www.orthomcl.org].
Martínez, J. Función e Importancia de la Bioinformática en el Desarrollo de las Ciencias, Especialmente en Biotecnología y MedicinaMolecular. Revisado Noviembre 2011. 2010.
Netbeans. Netbeans. Retrieved Diciembre de 2011. 2009 [Consultado el: 20 de diciembre de 2011]. Disponible en: [www netbeans.org].
Oxford, U. OrthologID. 2011. Disponible en: [http://bioinformatics.oxfordjournals.org/cgi/pmidlookup?view=long&pmid=16410324] : Bioinformatics 22 (6): pp. 699–707. doi:10.1093/bioinformatics/btk040. PMID 16410324.
Oxford, U. GreenPhylDB. 2012. Disponible en: [http://nar.oxfordjournals.org/cgi/pmidlookup?view=long&pmid=17986457] : Nucleic Acids Res. 36 (Database issue): pp. D991–8. doi:10.1093/nar/gkm934.
Pinna, M. (1991). "Conceptos y Análisis de homología en el Paradigma cladístico" . Cladistics 7 (4): p. 367-394.
Rumbaugh, J., Jacobson, I., & Booch., G. Unified Modeling Language Reference Manual. The Pearson Higher Education. 2004.
Schmitt, T. Letter to the Editor: SeqXML and OrthoXML: standards for sequence and orthology in-formation. Briefings in Bioinformatics. 2011.
Recibido: 27/02/2013
Aceptado: 25/06/2013


{kind=link}
{kind=link}
{kind=link}