










My SciELO
 Custom services
Custom servicesServices on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista Cubana de Ciencias Informáticas
On-line version ISSN 2227-1899
Rev cuba cienc informat vol.11 no.3 La Habana July.-Sept. 2017
ARTÍCULO ORIGINAL
Sistema para la extracción de información de proteínas y péptidos
Information retrieval system for proteins and peptides
Cosme E. Santiesteban-Toca1*, Liuben López Aparicio1
1Centro de Bioplantas, Universidad Máximo Gómez Báez, Ciego de Ávila, Carretera a Morón, Km 9½, Cuba, {liuben, cosme}@bioplantas.cu
*Autor para la correspondencia: cosme@bioplantas.cu
RESUMEN
El auge de las investigaciones en el área de las proteínas ha generado el desarrollo de grandes bases de datos en línea. El principal inconveniente de estas bases de datos es la descarga individual de las proteínas, lo cual se acrecienta con la dimensión de la búsqueda. Sin embargo, no se cuenta con una herramienta que extienda las funcionalidades de buscadores como PDBSelect, al análisis de las cadenas peptídicas y permita trabajar de forma conectada o desconectada al PDB. Por lo que en este artículo se presenta una herramienta que permite la búsqueda en cadenas peptídicas dentro del set de proteínas, así como de homologías entre proteínas y péptidos. Esta nueva herramienta facilita la selección y criba de proteínas, entre otras funciones de gran interés. Extendiendo las funcionalidades de PDBSelect, al permitir la búsqueda en cadenas peptídicas dentro del set de proteínas, así como de homologías entre proteínas y péptidos. La herramienta propuesta permite el trabajo de forma conectada o desconectada al PDB. El análisis de un caso de estudio permitió demostrar cómo, en apenas seis pasos sencillos, se logra obtener un conjunto de proteínas y subsecuencias peptídicas que permitan a los investigadores realizar posteriores investigaciones con estas.
Palabras clave: Herramienta bioinformática, búsqueda, péptidos, proteínas, alineamiento de secuencias, base de datos de proteínas.
ABSTRACT
The rise of research in the area of proteins has led to the development of large online databases. The main drawback of these databases is the download of individual proteins, which increases with the size of the search. However, we don´t have tools capable to extend the functionalities of search engines like PDBSelect, with respect to peptidical chains analysis and allowing to work both online or offline. In this paper, we present a tool that enables the search of peptidical chains inside the set of proteins and homologies between proteins and peptides. This tool facilitates the selection and screening of protein, among other features of interest. Extending PDBSelect functionality by allowing the search of peptide chains within the set of proteins and search homologies between proteins and peptides. The tool can work online or offline with the PDB. The analysis of a case of study helped to show how, in just six easy steps, it is possible to obtain a set of protein and peptide subsequences that allow researchers to conduct further research with these.
Key words: Bioinformatics tool, search, peptides, proteins, sequences alignment, proteins database.
INTRODUCCIÓN
El desarrollo alcanzado por las Ciencias Biológicas ha permitido la acumulación de gran cantidad de información experimental disponible en vastas bases de datos. El manejo e interpretación de estos volúmenes de información ha dado lugar al surgimiento de la Bioinformática.
En la actualidad existe una base de datos en línea de estructuras de proteínas y ácidos nucleicos nombrada Protein Data Bank (PDB) (Bi et al. 2015). Esta base de datos cuenta con aproximadamente 65 mil proteínas de estructura conocida. Cuyo objetivo fundamental es mantener un archivo de datos de estructuras macromoleculares disponible gratuita y públicamente para la comunidad global.
Existen múltiples bibliotecas y herramientas bioinformáticas ampliamente difundidas, destinadas al trabajo y la representación de estructuras de proteínas (Hirsh et al. 2015), (Colaert et al. 2013), (Sormanni et al. 2014), (Marchler-bauer et al. 2013), (Arnold, Bordoli y Schwede 2006), (Chandonia 2007), (Wu et al. 2007), (Stern et al. 2014) y (Wolstencroft et al. 2013). En sentido general estas bibliotecas y herramientas están diseñadas para el trabajo con una proteína a la vez o para realizar los alineamientos tradicionales empleados, locales o globales, sin atender a las relaciones que pudieran existir entre los péptidos de dichas proteínas y la actividad asociada a los mismos.
De igual manera existen múltiples bases de datos bien conocidas como: Protein Data Bank (Bi et al. 2015), (Consortium 2017), (Kiefer et al. 2009), (Hulo, N.; Amos, Bairoch; Bulliard, Virginie ; Cerutti, Lorenzo; De Castro, Edouard; Langendijk-Genevaux, Petra S.; Pagni, Marco; Sigrist 2006), (Mitchell et al. 2015). Todas estas bases de datos se encuentran en línea (on-line) y brindan potentes herramientas de búsqueda, que son capaces de filtrar por nombre, clasificación, características químicas, motivos estructurales, función, grado de identidad, organismos, etcétera. Sin embargo, tampoco están orientadas a la búsqueda de relaciones que pudieran existir entre los péptidos de dichas proteínas, ni permiten el trabajo desconectado (off-line).
El sitio de PDB posee una potente herramienta de búsqueda denominada PDBSelect, la cual tiene como principal inconveniente la descarga individual de las proteínas, lo cual se acrecienta con la dimensión de la búsqueda. Sin embargo, no se cuenta con una herramienta que extienda las funcionalidades de buscadores como PDBSelect, al análisis de las cadenas peptídicas y permita trabajar de forma conectada o desconectada al PDB.
Como parte del objetivo fundamental de la presente investigación, la herramienta deberá permitir la búsqueda en cadenas peptídicas dentro del set de proteínas, así como de homologías entre proteínas y péptidos.
Como resultado se propone una nueva herramienta que facilita la selección y criba de proteínas y péptidos, entre otras funciones de gran interés. Este artículo está conformado por una sección de antecedentes, donde se analizan algunas de las herramientas bioinformáticas empleadas en la selección y criba de proteínas. La sección donde se muestra la propuesta realizada. Una sección de validación de los resultados donde se muestra un caso de estudio y por ultimo las conclusiones y trabajos futuros.
MATERIALES Y MÉTODOS
En el presente artículo se propone una herramienta que facilita la investigación sobre la actividad que pueda tener una proteína o una determinada subsecuencia peptídica. Para ello se requiere que la herramienta creada sea capaz de encontrar las subsecuencias que se repiten en diferentes proteínas y la relación que existe entre la actividad de estas.
Esta herramienta brinda a los científicos e investigadores la posibilidad de interactuar directa o indirectamente con las bases de datos de proteínas en busca de posibles subsecuencias peptídicas para su posterior síntesis y análisis.
El Sistema Java de Recuperación de Información de Proteínas y Péptidos (JIRP, por sus siglas en ingles), está basado en la biblioteca de clases Bioinformatics, creada con el objetivo de extraer la información de las proteínas contenidas en el PDB, la realización de las búsquedas por subsecuencias peptídicas, así como las interfaces necesarias para lograr la interoperabilidad entre las bases de datos de proteínas, las herramientas bioinformáticas y los investigadores (Figura 1).
En este proceso se obtienen los genes con valores más centralizados y se eliminan los grupos con valores extremos. Con los genes resultantes tras este proceso de selección se construye un nuevo microarray que será sobre el que el algoritmo de inferencia realice la extracción de las redes de genes.
Implementación
JIRP es una herramienta de escritorio desarrollada con Java y libre que permite organizar el trabajo de investigación asociado al manejo de proteínas. Esta herramienta ofrece un ambiente gráfico para el diseño de búsqueda altamente personalizada de proteínas y de subsecuencias en las mismas.
La aplicación cuenta con una base de datos interna, que viabiliza el proceso de búsqueda de y dentro de las proteínas. Esto se logra a partir de la extracción de las propiedades de las proteínas derivadas tanto de su estructura primaria, secundaria como terciarias. Además, almacena las subsecuencias y sus coincidencias, creando así una base de datos de plantillas de péptidos asociados a sus posibles actividades.
El sistema es capaz de leer proteínas en formato *.pdb y *.ent, las cuales pudieran encontrarse en ficheros compactados *.gz. La búsqueda de proteínas se realiza empleando los mismos criterios de PDBSelect, extendiendo estos a esquemas de estructuras secundarias definidas por el investigador. Estas búsquedas también se pueden realizar a partir de una subsecuencias de aminoácidos previamente guardada por el investigador en la base de datos, devolviéndole secuencias que tengan la misma función o similar a la que se utiliza como plantilla para la búsqueda. La búsqueda de secuencias también se realiza según criterios de hidrofobicidad en aminoácidos presentes en dichas secuencias, así como aminoácidos deseados o no.
La búsqueda a través de estructuras secundarias definidas por el investigador se simplifica a que plantee exactamente la forma de la subsecuencia que desea encontrar en otras proteínas, esta se trasforma en tiempo de ejecución a una expresión regular que se encarga de realizar las comparaciones en la base de datos, teniendo como resultado las proteínas coincidentes y similares. De esta manera los investigadores no necesitan de los conocimientos de informática para tener éxito.
Como complemento a este método de búsqueda se encuentra un motor de sugerencias basado en los posibles errores del investigador en el momento de construir la expresión.
Otra característica importante del proceso de búsqueda es el empleo de plantillas. Las cuales se basan en búsquedas previas que fueron guardadas en la base de datos y cuya subsecuencia está asociada a una actividad bien definida. El empleo de dichas plantillas está pensado en base a la eficiencia de las búsquedas debido a que funcionan como un tipo de cache.
El uso de este método tiene dos aristas fundamentales porque de no encontrarse la plantilla adecuada se tendría que proceder a realizar un parseo en toda la base de datos, lo cual se evitaría de aparecer, reduciendo notablemente el tiempo de búsqueda.
JIRP permite realizar alineamientos locales y globales lo que facilita la comparación de secuencias de proteínas resaltando las zonas de similitud que podrían indicar relaciones funcionales o evolutivas entre las proteínas seleccionadas.
A diferencia de las herramientas tradicionales, JIRP permite exportar las proteínas seleccionadas, por cualquiera de los métodos anteriormente descritos, hacia una carpeta o una nueva base de datos para realizar experimentación posterior con ellas. En caso que desee exportarlas en forma de fichero el sistema busca la dirección de procedencia de las proteínas e intenta copiarla desde esta o las extrae directamente de la base de datos si desea exportar a una base de datos nueva.
Base de datos
La aplicación cuenta con una base de datos interna SQLite. En esta base de datos se gestionan una serie de índices derivados de los metadatos extraídos de los pdb que permiten al investigador crear criterios de búsqueda personalizados y obtener la información organizada según su relevancia y cobertura.
A medida que la aplicación indexa nuevas proteínas la información es almacenada en tablas a modo de relaciones, tal que permita la recuperación desde diferentes perspectivas como actividad, estructura, función, familia (Figura 2).
En la medida en que se introducen nuevas subsecuencias peptídicas asociadas a una actividad específica, se crean las relaciones entre estas y las proteínas que las contengan. Esto se pone de manifiesto con mucha frecuencia debido a que una subsecuencia peptídica puede estar en varias proteínas o varias veces en una misma proteína, donde pueden encontrarse separadas o solapadas. A partir de la relevancia y cobertura se realiza la organización de la devolución que da la base de datos ante determinada búsqueda a partir de una subsecuencia peptídica. El trabajo offline se puede realizar debido a que la base de datos interna de la aplicación está indexada con PDB. La importación a la base de datos está flexibilizada por la variabilidad de los ficheros pdb.
Diseño de la interfaz del sistema
La interfaz de la aplicación está dividida en 5 áreas fundamentales (Figura 3):
-
En la barra de herramientas están contenidas las funciones básicas de la aplicación y el acceso al resto de las paletas.
-
En la paleta de gestión de base de datos se encuentran las bases de datos que han sido cargadas o creadas recientemente.
-
El área de visualización de las proteínas es donde se muestran las proteínas que han sido seleccionadas a partir de una búsqueda previa.
-
La paleta de búsquedas se compone de dos subpaletas que contienen dos tipos de búsqueda diferentes (búsqueda de proteínas y búsqueda de subsecuencias) a las que se puede acceder mediante la barra de herramientas.
-
La barra de estado muestra a la izquierda la cantidad de proteínas que han sido seleccionadas producto de una búsqueda y a la derecha si se están cargando nuevas proteínas para la base de datos y el porciento en que se encuentra este proceso.
RESULTADOS Y DISCUSIÓN
Para analizar el funcionamiento de la herramienta propuesta, se diseñó un caso de estudio. Donde, se desea crear un modelo que sea capaz de predecir actividad antimicrobiana de péptidos. Una primera aproximación podría ser realizar una búsqueda entre los segmentos de proteínas conocidas (PDB). Debido a que los péptidos de varios tipos naturales como piel de anfibios, de arañas y péptidos propios del ser humano son antimicrobianos que exhiben patrones de secuencias, una idea de partida pudiera ser buscar en proteínas esos patrones y sacar estos fragmentos para posterior-mente realizar experimentación con ellas.
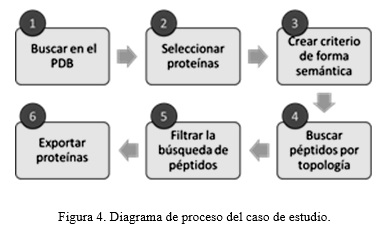
En la figura 4 se muestran los pasos que se siguen en este proceso. Primero, se realiza la búsqueda de proteínas en el PDB. Segundo, se selecciona el conjunto de proteínas objetivo, pueden ser todas o solo un subconjunto de la búsqueda. Tercero, se establece el criterio de búsqueda de forma semántica. Cuarto, búsqueda de péptidos según estructura 2D. Quinto, se refina la búsqueda atendiendo a las características físico químicas de los aminoácidos. Por último, se exportar las proteínas que cumplen con los criterios anteriores.
Para la realización de la búsqueda inicial de proteínas en el PDB, se establecieron los siguientes parámetros (Figura 5.2): secuencias con longitud de más de 50 aminoácidos, resolución mayor que 65, obtenidas por difracción de rayos X y organismos de procedencia de anfibios.
Como resultado del paso uno y dos del proceso, se obtiene un grupo de proteínas que se supone puedan estar relacionadas con la actividad buscada (Figura 5.1).
Basándonos en la afirmación de que la estructura secundaria de los péptidos antimicrobianos de tipo uno presenta alfa hélices (Jenssen, Hamill y Hancock 2006), el tercer paso es realizar una criba de péptidos que contengan este motivo 30 veces.
Para realizar esta búsqueda la expresión semántica sería “HHHHHHH…HH” hasta llegar a 30. Sin embargo, la construcción de la expresión de búsqueda está dada por el motivo, seguido del número de veces en que debe estar repetido. Esto provoca que ejecute el motor de sugerencias de expresiones con H30, lo cual genera la expresión regular H{30}. La figura 6.1 muestra los segmentos de alfa hélice que coincidieron con la expresión regular y las secuencias de aminoácidos asociadas a los mismos.
En la mayoría de los casos la búsqueda realizada en los pasos tres y cuatro genera una gran cantidad de subsecuencias peptídicas, por los que podría hacerse necesario refinar esta búsqueda. En el quinto paso se acota la búsqueda mediante el empleo de filtros. Como especificar si se permite que las subsecuencias puedan solaparse (en este caso se permitió hasta 5 aminoácidos de solapamiento). Así como especificar los aminoácidos que puedan estar presentes y los que no (en este caso debían estar presentes Prolina y Glicina, y no la Cisteína). Adicionalmente los aminoácidos pueden elegirse según los criterios de hidrofobicidad, polaridad y carga (Figura 6.2).
Los péptidos que cumplen con las restricciones anteriores no siempre se parecen entre sí, por lo que se hace necesario encontrar las secuencias más similares. Estas secuencias son asociadas con la actividad antimicrobiana analizada y se almacenan en la base de datos con referencia a sus respectivas proteínas ( figura 6.1). Estas subsecuencias servirán como plantilla para búsquedas posteriores.
Como último paso, las proteínas que contienen a los péptidos seleccionados pueden ser exportadas hacia una carpeta tal que se puedan realizar experimentaciones específicas con estas.
CONCLUSIONES
Como resultado se obtuvo una nueva herramienta que facilita la selección y criba de proteínas, entre otras funciones de gran interés. Extendiendo las funcionalidades de PDBSelect, al permitir la búsqueda en cadenas peptídicas dentro del set de proteínas, así como de homologías entre proteínas y péptidos. La cual permite el trabajo de forma conectada o desconectada al PDB.
JIRP es una herramienta portable, desarrollada en Java, que consume pocos recursos y brinda una interfaz simplificada e intuitiva para los investigadores. El empleo de un caso de estudio que permitió demostrar seis pasos sencillos la forma de obtener un conjunto de proteínas y subsecuencias peptídicas que permitan a los investigadores realizar posteriores investigaciones con estas.
AGRADECIMIENTOS
Al Laboratorio de Bioinformática del Centro de Bioplantas, en Ciego de Ávila, Cuba.
REFERENCIAS BIBLIOGRÁFICAS
ARNOLD, K., BORDOLI, L. y SCHWEDE, T., 2006. Structural bioinformatics The SWISS-MODEL workspace : a web-based environment for protein structure homology modelling. Bioinformatics, vol. 22, no. 2, pp. 195-201.
BI, C., BLUHM, W.F., CHRISTIE, C.H., ROSE, P.W., PRLI, A., DUTTA, S., GREEN, R.K., GOODSELL, D.S., WESTBROOK, J.D., WOO, J., YOUNG, J., ZARDECKI, C., BERMAN, H.M., BOURNE, P.E., RCSB, T., DATA, P. y RCSB, B., 2015. The RCSB Protein Data Bank : views of structural biology for basic and applied research and education. Nucleic Acids Research, vol. 43, no. November 2014, pp. 345-356.
CHANDONIA, J., 2007. Structural bioinformatics StrBioLib : a Java library for development of custom computational structural biology applications. Bioinformatics, vol. 23, no. 15, pp. 2018-2020.
COLAERT, N., MADDELEIN, D., IMPENS, F., DAMME, P. Van, GEVAERT, K. y MARTENS, L., 2013. The Online Protein Processing Resource ( TOPPR ): a database and analysis platform for protein processing events. Nucleic Acids Research, vol. 41, no. October 2012, pp. 333-337.
CONSORTIUM, T.U., 2017. UniProt : the universal protein knowledgebase. Nucleic Acids Research, vol. 45, no. November 2016, pp. 158-169.
HIRSH, L., PIOVESAN, D., GIOLLO, M., FERRARI, C. y TOSATTO, S.C.E., 2015. Structural bioinformatics The Victor C11 library for protein representation and advanced manipulation. Bioinformatics, vol. 31, no. November 2014, pp. 1138-1140.
HULO, N.; AMOS, BAIROCH; BULLIARD, VIRGINIE ; CERUTTI, LORENZO; DE CASTRO, EDOUARD; LANGENDIJK-GENEVAUX, PETRA S.; PAGNI, MARCO; SIGRIST, C.J.A., 2006. The PROSITE database. Nucleic Acids Research [en línea], vol. 34, no. 90001, pp. D227-D230. ISSN 0305-1048. DOI 10.1093/nar/gkj063. Disponible en: https://academic.oup.com/nar/article-lookup/doi/10.1093/nar/gkj063.
JENSSEN, H., HAMILL, P. y HANCOCK, R.E.W., 2006. Peptide antimicrobial agents. Clinical Microbiology Reviews, vol. 19, no. 3, pp. 491-511. ISSN 08938512. DOI 10.1128/CMR.00056-05.
KIEFER, F., ARNOLD, K., KÜNZLI, M., BORDOLI, L. y SCHWEDE, T., 2009. The SWISS-MODEL Repository and associated resources. Nucleic Acids Research, vol. 37, no. SUPPL. 1, pp. 387-392. ISSN 03051048. DOI 10.1093/nar/gkn750.
MARCHLER-BAUER, A., ZHENG, C., CHITSAZ, F., DERBYSHIRE, M.K., GEER, L.Y., GEER, R.C., GONZALES, N.R., GWADZ, M., HURWITZ, D.I., LANCZYCKI, C.J., LU, F., LU, S.,
MARCHLER, G.H., SONG, J.S., THANKI, N., YAMASHITA, R.A., ZHANG, D. y BRYANT, S.H., 2013. CDD : conserved domains and protein three-dimensional structure. Nucleic Acids Research, vol. 41, no. November 2012, pp. 348-352.
MITCHELL, A., CHANG, H.Y., DAUGHERTY, L., FRASER, M., HUNTER, S., LOPEZ, R., MCANULLA, C., MCMENAMIN, C., NUKA, G., PESSEAT, S., SANGRADOR-VEGAS, A., SCHEREMETJEW, M., RATO, C., YONG, S.Y., BATEMAN, A., PUNTA, M., ATTWOOD, T.K., SIGRIST, C.J.A., REDASCHI, N., RIVOIRE, C., XENARIOS, I., KAHN, D., GUYOT, D., BORK, P.,
LETUNIC, I., GOUGH, J., OATES, M., HAFT, D., HUANG, H., NATALE, D.A., WU, C.H., ORENGO, C., SILLITOE, I., MI, H., THOMAS, P.D. y FINN, R.D., 2015. The InterPro protein families database: The classification resource after 15 years. Nucleic Acids Research, vol. 43, no. D1, pp. D213-D221. ISSN 13624962. DOI 10.1093/nar/gku1243.
SORMANNI, P., CAMILLONI, C., FARISELLI, P. y VENDRUSCOLO, M., 2014. Simultaneous Sequence-Based Prediction of the Statistical Populations of Ordered and Disordered Regions in Proteins. Journal of Molecular Biology, vol. 43, pp. 345-356.
STERN, P., HÅKANSSON, A., TÄHTINEN, M. y ANGELIS, J., 2014. Evaluation of the NeoBio and SymBio programmes. Tekes,
WOLSTENCROFT, K., HAINES, R., FELLOWS, D., WILLIAMS, A., WITHERS, D., OWEN, S., SOILAND-REYES, S., DUNLOP, I., NENADIC, A., FISHER, P., BHAGAT, J., BELHAJJAME, K., BACALL, F., HARDISTY, A., NIEVA, A., HIDALGA, D., VARGAS, M.P.B., SUFI, S. y GOBLE, C., 2013. The Taverna workflow suite : designing and executing workflows of Web Services on the desktop , web or in the cloud. Nucleic Acids Research, vol. 41, no. May, pp. 557-561.
WU, D., CUI, F., JERNIGAN, R. y WU, Z., 2007. PIDD : database for Protein Inter-atomic Distance Distributions. Nucleic Acids Research, vol. 35, no. December 2006, pp. 202-207.
Recibido: 20/05/2017
Aceptado: 25/06/2017

