










 Servicios personalizados
Servicios personalizados
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
Los microarreglos son en la actualidad una poderosa herramienta de análisis de expresión de genes debido al mayor número de secuencias que se pueden analizar por prueba, constituyendo una ventaja con respecto al resto de las técnicas convencionales, en las cuales solo se pueden analizar un número limitado de genes de manera simultánea, debido a que en la mayoría de los casos se requiere montar un experimento por cada gen a estudiar. De esta forma, existen microarreglos tanto de proteínas como de ADN(Allanki & Dixit, 2017).
Un ensayo de un microarreglo para diagnóstico, es una técnica que consiste en la adsorción de biomoléculas sobre un soporte sólido formando una matriz. Dichas biomoléculas se incuban con la muestra a analizar, donde se produce el reconocimiento y unión con las moléculas complementarias (Salem & Bahari, 2017). A continuación, la reacción es revelada con el uso de marcadores fluorescentes, generalmente de cianina, Cy3 (rojo) y Cy5 (verde), los cuales tienen emisiones en los rangos de 510-550 nm y 630-660 nm, respectivamente. Posteriormente, se obtiene la imagen a través de un lector y las herramientas bioinformáticas permiten interpretar y analizar los resultados(Binti, 2015).
Para un típico procedimiento, se producen dos archivos de imagen, uno para cada tinte fluorescente. Estas imágenes son los datos crudos del experimento. Por tanto, se hace necesario un correcto análisis de las mismas, el cual consta de tres etapas: pre- procesamiento y grillado, segmentación y extracción de características (Salem & Bahari, 2017) (Shao, Li et al., 2019) (R.M. Farouk & SayedElahl, 2019).
En aras de aprovechar las potencialidades que brinda la tecnología de microarreglos de ADN para diagnóstico, en el centro de Inmunoensayo (CIE), ubicado en La Habana, Cuba, se desarrolla la Tecnología Suma (Sistema Ultra Micro Analítico) como parte indispensable de las estrategias del Ministerio de Salud Pública para brindar métodos diagnósticos con la mejor calidad a un costo mínimo y proporcionar una herramienta ideal para los programas de salud basados en pesquisajes masivos.
Entre los programas de Pesquisa Neonatal que se desarrollan en el centro, se encuentra un proyecto encaminado al Diagnóstico molecular de la Fibrosis Quística (FQ). Esta es una enfermedad causada por una mutación en un gen llamado regulador de la conductancia transmembrana de la fibrosis quística (CFTR, por sus siglas en inglés), que afecta al organismo de forma generalizada causando muerte prematura(Galvina, Clarkeb, Harveya, & Amaralb, 2004).
Esta es una enfermedad hereditaria autosómica recesiva, es decir, que los pacientes pueden tener dos mutaciones: una del alelo paterno y otra del alelo materno. De acuerdo a cómo se presentan estas mutaciones, existen las siguientes clasificaciones: portador sano o paciente heterocigótico: un solo alelo mutado, heterocigóticos compuestos: dos mutaciones diferentes, una en cada parental, por lo tanto es un paciente enfermo y por último, paciente homocigótico: contiene la misma mutación en los dos parentales y no se expresa este gen de forma normal(Armas et al., 2019).
La prueba del sudor constituye el estándar de oro para el diagnóstico confirmatorio de FQ, mientras que el análisis genético se utiliza en la detección prenatal y preconcepcional de portadores con el fin de identificar el riesgo de tener un hijo con FQ. Teniendo en cuenta su utilidad el análisis genético debería incluirse en el diagnóstico de rutina(Armas et al., 2019).
Para que nuestro sistema de salud cubano disponga de este ensayo se necesita realizar procesos de síntesis de oligonucleótidos, placas Greiner, un robot dispensador de microarreglos y un lector con LEDs de alto brillo para la excitación de los microarreglos de los oligonucleótidos dispensados.
El logro de todos estos elementos facilitaría la detección temprana de los niños que presentan esta enfermedad, incidiendo en la disminución de la mortalidad de estos y permitiría obtener una tecnología propia del país que nos dotaría de independencia y sería más económica pues en el mercado internacional, un test (ADN) de FQ para una persona ronda los 900 dólares.
Por tales motivos, el siguiente trabajo presenta el software desarrollado con la finalidad de automatizar el lector de Microarreglos con LEDs de alto brillo, diseñado en el CIE, que permitirá capturar y procesar las imágenes de microarreglos de oligonucleótidos, resultantes de los ensayos para el diagnóstico de la FQ. Este software reconocerá las mutaciones del gen CFTR y logrará diferenciar entre el alelo normal (marcado con Cy5) y el mutado (marcado con Cy3) detectando si los pacientes son portadores sanos, heterocigóticos compuestos u homocigóticos.
MATERIALES Y MÉTODOS
El procesamiento de las imágenes de microarreglos constituye el punto de partida para el posterior análisis biológico de los datos. Por tal motivo, es necesario realizar su implementación en una aplicación computacional que contenga las tres etapas fundamentales para el análisis de las imágenes.
Primeramente, la etapa de pre- procesamiento y grillado realiza un algoritmo inicial con la implementación de filtros, operaciones morfológicas, normalización, etc., que permite reducir los ruidos y enriquecer la calidad de la imagen(González, 2010). A continuación, se ejecuta el proceso de grillado que consiste en la asignación de coordenadas a cada uno de los puntos en la imagen. Este procedimiento puede ser de forma manual, semi-automática o automática. Muchos artículos han sido publicados presentando diferentes técnicas de grillado (Rueda y Rezaeian, 2011) (Ahmad, Bahari, & Yusoff, 2014).
En esta investigación, se implementa un método de grillado que se basa fundamentalmente en el cálculo de la proyección horizontal y vertical de la imagen, así como el centro de masa de sus puntos (Binti, 2015).
Otro elemento importante y de los más desafiantes lo constituye la segmentación de las imágenes como procedimiento para agrupar los pixeles con características similares, separando los pixeles considerados como señales de aquellos que son fondo en las imágenes(Torres, 2014).
Existen diferentes métodos de segmentación que pueden ser categorizados en los siguientes grupos principales de acuerdo a la geometría de los puntos que ellos producen:
Círculo fijo o adaptable: Estos métodos son fáciles de implementar pero funcionan bien si todos los puntos son circulares y del mismo tamaño(Fouad, Mabrouk , & Sharawy, 2014).
Métodos basados en umbral: Estos métodos pueden detectar con más precisión los puntos del microarreglo debido a que se adapta a sus formas reales. Ejemplo de ellos son: Crecimiento de regiones, método de umbralización global y local, Marcov Random Field, etc(R.M. Farouk & SayedElahl, 2019).
Reconocimiento de patrones: Estos métodos se dividen en aprendizaje supervisado y no supervisado, y se basan en alguna medida de similitud entre los pixeles parar realizar el agrupamiento. Son técnicas de segmentación más comunes que tienen la ventaja de no estar restringidas a una forma o tamaño particular de los puntos dentro de la imagen. Entre ellos se pueden citar: Máquinas de soporte vectorial, Redes Neuronales Artificiales, agrupamiento de K-medias, Media de Fuzzy C y Expectación-Maximización(Shao, Li, Zuo, Wu, & Liu, 2015).
Por último, la extracción de características tiene el propósito de obtener los niveles de expresión de los genes de acuerdo a los resultados obtenidos en los procesos anteriores. Es conveniente encontrar métodos de extracción de características que minimicen la pérdida de información(Wu, A. Su, & Billings, 2012).
METODOLOGÍA COMPUTACIONAL
En una primera etapa de la investigación se estudiaron e implementaron varios algoritmos para analizar una imagen de microarreglos para diagnóstico, pasando por las tres etapas del análisis de la imagen.
Estas implementaciones (Figura 1) forman parte del software desarrollado para la comunicación con el Lector de Microarreglos con LEDs de alto brillo, diseñado con el objetivo de brindar una herramienta para la interpretación de los resultados por parte de los analistas y de los laboratorios donde se implemente la tecnología.
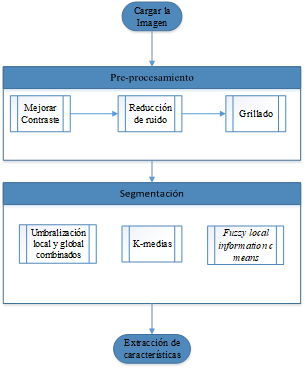
Una vez obtenida la imagen y separada en los dos canales (Figura 2), en la etapa de pre-procesamiento, se procede a realizar un ajuste de contraste para realzar la calidad de la imagen y ajustar sus valores de intensidad. Posteriormente, se aplica un filtro de mediana para reducir en las imágenes el ruido resultante de errores durante el proceso experimental.

A continuación, se realiza el proceso de grillado cuyo rol principal es dividir los puntos del microarreglo en áreas independientes. El siguiente algoritmo (Figura 3) ilustra su implementación de forma automática:
Obtener la proyección horizontal y vertical de la imagen, a partir de (ec.1, 2) donde g(x,y) es la imagen proveniente del filtrado anterior.
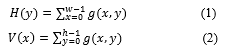
Remover morfológicamente, a través de la transformación top_hat, el fondo de la intensidad de cada proyección.
Localizar y segmentar los picos de cada región por umbralización.
Determinar los centros de masa y la distancia entre los puntos.
Obtener las coordenadas de ambos ejes así como el número de líneas para el grillado.
Posicionar la rejilla conformada sobre la imagen original
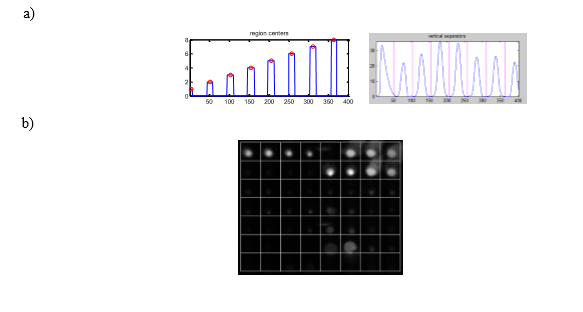
Fig.3 a) Cálculo de los centros de masa y la distancia entre puntos en la proyección vertical en una imagen de microarreglo b) Imagen resultante del pre-procesamiento
La exactitud del método de grillado desarrollado en una imagen, teniendo el número total de puntos, puede ser calculado como:
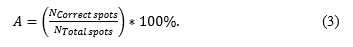
Donde N Correct spots , 𝑁 𝑇𝑜𝑡𝑎𝑙 𝑠𝑝𝑜𝑡𝑠 indican el número de puntos correctamente grillados y el total de puntos en la imagen respectivamente (Fouad et al., 2014).
En la etapa de segmentación se implementaron tres algoritmos y se deja a libre elección por parte del usuario cual es el más óptimo a utilizar según las necesidades presentes Figura 4:
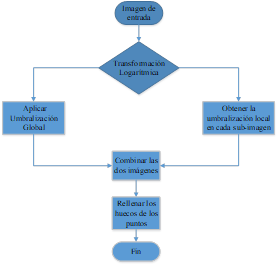
Método por umbralización local y global combinados: El primer paso es ecualizar las diferencias de iluminación entre los puntos con la aplicación de una transformación logarítmica. A continuación se realiza la umbralización global sobre toda la imagen, la cual permite separar intensidades en dos picos en la imagen. La umbralización local se realiza sobre sub-imágenes y se determina un umbral para cada partición. Posteriormente, se combinan ambas imágenes, lo cual permite linealizar la intensidad de los puntos, obteniéndose mejores resultados. Además, se rellenan los huecos en los puntos para evitar la unión de unos con otros o mala detección de sus bordes(Fouad et al., 2014).
Agrupamiento por K-medias: Los algoritmos de agrupamiento requieren de la inicialización de los centroides de los clústeres (SivaLakshmi & Malleswara, 2018), que en el caso de imágenes de microarreglos para diagnóstico serán sólo dos: Señal y Fondo. Mientras más representativos sean estos valores iniciales más rápido se obtendrá la convergencia de los algoritmos. Se propone considerar el píxel de mínimo valor de intensidad, como el representante del clúster que estará formado por los píxeles que clasificaremos como pertenecientes al Fondo y utilizar el píxel de máximo valor de intensidad, como representante del clúster que estará formado por los píxeles que clasificaremos como pertenecientes a la Señal. Esta propuesta es debido a que en la metodología del dispensado de las matrices de microarreglos, en los centros de cada punto estará siempre el píxel de mayor valor de intensidad.
En el caso de K-medias, se necesita además, establecer una medida de similitud o distancia a través de la función criterio que se muestra en la (ec.4) donde 𝑢 𝑗 es la media del clúster designado(Uslan & Bucak, 2010).
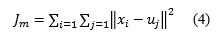
El algoritmo es el siguiente:
Escoger K puntos como representantes de los clústeres iniciales.
Asignar cada dato al clúster más cercano, con el sentido de minimizar la diferencia entre el valor del dato y el centroide del clúster.
Recalcular la posición de los K centroides, una vez asignados los datos.
Repetir 2 y 3 hasta que todos los datos hayan sido clasificados.
Un valor absoluto de la diferencia entre dos funciones criterios consecutivas en este método, 𝐽 𝑚+1 y 𝐽 𝑚 , es el elemento buscado para minimizar iterativamente hasta la condición de parada, siendo menor que un parámetro εk determinado.
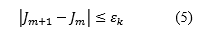
Fuzzy local information c means (FLICM): Esta técnica es una versión modificada del Fuzzy c means, la cual utiliza una medida de similaridad fuzzy local, con el objetivo de garantizar la insensibilidad al ruido y la preservación de los detalles de la imagen. En este sentido, se adiciona un nuevo factor 𝐺 𝑖𝑗 (ec.6) en la función objetivo del Fuzzy c means. Este factor incorpora un nivel local de grises y una información espacial local, controlando la influencia de los pixeles vecinos dependiendo de la distancia de estos con el pixel central.
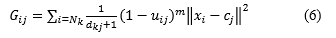
Donde el pixel 𝑘 𝑡ℎ es el centro de la ventana local, j es el cluster de referencia y el pixel ?? 𝑡ℎ pertenece al grupo de vecinos en la ventana alrededor del 𝑘 𝑡ℎ pixel 𝑁 𝑘 . 𝑑 𝑘𝑗 es la distancia Euclidiana entre los pixeles k y j, 𝑢 𝑖𝑗 (ec.7) es el grado de membresía del pixel 𝑖 𝑡ℎ en el cluster 𝑗 𝑡ℎ , m es el exponente de peso de cada membresía y 𝑐 𝑗 es el centro del cluster j (ec.8)(V.G. & P., 2014).
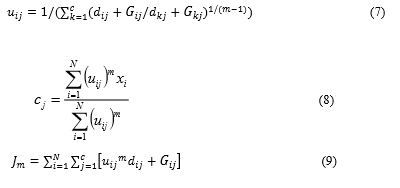
Algoritmo:
Asignar valores iniciales a los centroides 𝑐 𝑗 de cada clúster, al parámetro m y fijar una condición de parada (.
Calcular 𝑢 𝑖𝑗 según (ec.7).
Calcular los centroides 𝑐 𝑗 según (ec.8).
Evaluar en la función objetivo con (ec.9).
Repetir los pasos del 2 al 4 hasta que se cumpla que:
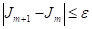 (10)
(10)
La expresión del gen en la imagen se obtiene en (ec.11) donde 𝐼 𝑐𝑦3 e 𝐼 𝑐𝑦5 corresponde a las intensidades de ambos canales (ec.12).
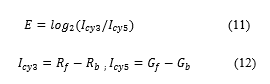
Donde R f , G f representan la intensidad media de los pixeles clasificados señal y R b , G b representan la intensidad media de los pixeles clasificados como fondo en la imagen.
Para realizar un análisis comparativo de los procesos de segmentación realizados, se evalúa el error cuadrático medio normalizado como medida cuantitativa (ec.13).
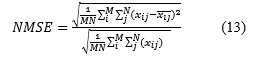
Donde M y N son las dimensiones de la imagen, 𝑥 𝑖𝑗 , 𝑥 𝑖𝑗 son los pixeles originales y de los clústeres respectivamente. Un valor cercano a cero de NMSE significa que la segmentación realizada presenta precisión, por lo cual es correcta.
RESULTADOS Y DISCUSIÓN
Los métodos presentados fueron puestos a prueba en un grupo de 38 imágenes de pacientes con FQ enviadas por la Comisión Nacional FQ. De esta forma, se pudieron comprobar cada una de las etapas, obteniéndose los siguientes resultados:
Se logró una exactitud del 96% en la fase de grillado en más del 50% de las imágenes analizadas.
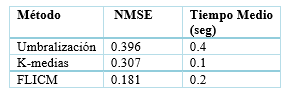
Los métodos de segmentación implementados arrojaron buenos resultados en su desempeño (Figura 5), destacándose el K-medias y el FLICM por tener los menores valores de NMSE y los menores tiempos de ejecución (Tabla 1), aunque la implementación de cualquiera de ellos permitió detectar las mutaciones presentes.

Fig.5 a) Punto original de una imagen de microarreglos b) segmentación por umbralización c) segmentación por K-medias. d) segmentación por FLICM
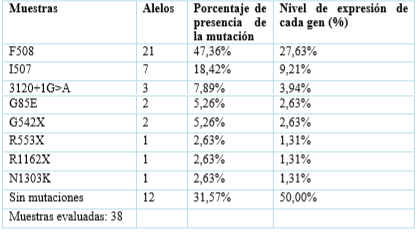
Finalmente, la etapa de extracción de características reconoció 8 tipos de mutaciones del gen CFTR y los niveles de expresión de las mismas. Además, se pudo concluir que de ellos el 47% presentan la mutación F508. El 50% de los alelos afectados no se pudieron determinar pues no se corresponden a ninguna de las 33 mutaciones, con alta frecuencia a nivel mundial, detectadas (Tabla 2).
Se puede concluir, que el software desarrollado permite a través de la ejecución de sus etapas, un procesamiento de las imágenes de microarreglos satisfactorio para su uso posterior por parte de los expertos. Se obtuvo la clasificación de los pacientes a través de la expresión de las mutaciones detectadas.
Por ejemplo, a partir de la comparación de una muestra normal marcada con Cy5 (Figura 6) con otras muestras mutadas marcadas con Cy3, se obtuvieron los siguientes resultados:
Portador sano con uno de sus alelos mutados con R334W y el otro con expresión normal (Figura 7 a)).
Paciente heterocigótico compuesto, el cual presenta dos mutaciones en sus parentales, 3120+1G>A y F508, presentándose estos mismos genes en la muestra normal (Figura 7 b)).
Paciente homocigótico debido a que contiene la misma mutación G542 en sus dos parentales, la cual no se expresa en la muestra normal (Figura 7 c)).


CONCLUSIONES
La herramienta computacional diseñada permite realizar un estudio para evaluar cualitativamente por fluorescencia las mutaciones del gen CFTR más frecuentes en Cuba y América Latina, así como determinar dentro de un microarreglo si hay más de una mutación para un paciente determinado. La misma, cuenta con tres algoritmos de segmentación para separar los puntos de máxima intensidad de aquellos que presentan la mínima intensidad y son considerados fondo en las muestras. Estos algoritmos pueden ser empleados indistintamente por los expertos debido a que la implementación de cada uno de ellos permitió detectar las mutaciones presentes en las imágenes procesadas.
Actualmente, este software ha sido incorporado al Lector de Microarreglos con Leds de alto brillo diseñado, dotando al equipo de potencialidades y ventajas con respecto a versiones anteriores, para el método de diagnóstico de varias enfermedades simultáneamente y para la realización del monitoreo de los estadios de las mismas.

