










 Custom services
Custom services
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
Permalink
INTRODUCCIÓN
La epidemiología molecular en enfermedades infecciosas se usa ampliamente para definir la fuente de infección, así como las relaciones ancestrales entre los patógenos aislados de los individuos muestreados de una población. La teoría coalescente y el análisis filogeográfico han tenido aplicación científica en varios eventos recientes de pandemia y brotes nosocomiales (Ciccozzi, Lai et al., 2019). Los datos sobre secuencias aisladas del microorganismo son esenciales para aplicar las herramientas filogenéticas y la investigación en el campo de la filodinámica de las enfermedades infecciosas está creciendo.
La propagación pandémica en curso de un nuevo coronavirus humano, el SARS-Cov-2, asociado con la enfermedad de neumonía grave (COVID-19), ha dado como resultado la generación de millones de secuencias de genomas de virus, con una tasa de generación de genomas sin precedentes (Rambaut, Holmes et al., 2020). En la base de datos EpiCovTM asociada a la iniciativa GISAID https://www.gisaid.org/ (Elbe and Buckland‐Merrett 2017; Shu and McCauley, 2017), y disponible en https://www.epicov.org/ bajo subscripción, se puede encontrar el principal reservorio de secuencias genómicas de SARS-Cov-2, así como los metadatos asociados a ellas.
Esta alta disponibilidad de secuencias genómicas ha favorecido la realización de estudios filogenéticos, filodinámicos y filogeográficos (en realidad filogenómicos, al emplear todo el genoma) que han aumentado considerablemente el conocimiento sobre el virus, permitiendo inferir su posible origen, su tasa evolutiva, las vías de distribución geográfica y el establecimiento de los primeros esfuerzos en su clasificación (Lu, Zhao et al., 2020; Lv, Li et al., 2020; Volz, Fu et al., 2020; Zhou, Yang et al., 2020).
Para comprender la evolución y transmisión del SARS-CoV-2, el genotipado de los aislados de virus es de gran importancia (Yin, 2020). A medida que el brote evoluciona con el tiempo y hay más datos disponibles, se han propuesto diferentes sistemas de clasificación de las variantes (Forster, Forster et al., 2020; Hodcroft, 2020; Rambaut, Holmes et al., 2020; Yin, 2020). Entre los sistemas de clasificación del SARS-Cov-2 basados en árboles, el de (Rambaut, Holmes et al., 2020), empleado por GISAID, propone un sistema dinámico para etiquetar linajes transitorios que tienen importancia epidemiológica local. Otro enfoque basado en árboles filogenéticos es el que ofrece la plataforma Nextstrain (Hadfield, Megill et al., 2018), en el que se introduce un sistema de nombres que facilita la discusión de los patrones de diversidad a gran escala del SARS-CoV-2 y las etiquetas de clados que persisten durante al menos varios meses y tienen una distribución geográfica significativa (Hodcroft, 2020).
Las limitaciones tecnológicas existentes, no permiten que en Cuba se realice la secuenciación del genoma completo de las cepas aisladas del SARS-Cov-2. No obstante, existe la capacidad para secuenciar pequeños fragmentos de genoma mediante tecnología Sanger, por lo que la selección de marcadores que reflejen el comportamiento del virus constituye un paso determinante en el proceso de estudio.
Antes de existir la posibilidad de la secuenciación completa de genomas, seleccionar el mejor o los mejores genes y el conjunto más informativo de taxones se convirtió en uno de los principales problemas en la inferencia filogenética (Nabhan and Sarkar, 2012). Entre las buenas cualidades de un marcador, está capacidad de ser utilizado para producir árboles filogenéticos robustos que reflejen tanto como sea posible la evolución de las especies de las cuales provienen los genes (Wu, Jospin et al., 2013). En un análisis filogenético molecular, diferentes marcadores pueden producir topologías contradictorias para el mismo grupo de diversidad. Cuestiones como la longitud y la tasa de evolución jugarán un papel en la idoneidad de un marcador molecular particular para desarrollar las relaciones filogenéticas para un conjunto dado de taxones combinado (Russo, Aguiar et al., 2017).
En el presente trabajo se realiza un estudio de las subregiones genómicas del SARS-Cov-2 para determinar su posible valor como marcadores filogenéticos. Se sigue como premisas que una subregión del genoma será un buen marcador filogenético para el SARS-Cov-2 si la señal filogenética es fuerte y permite reproducir el historial evolutivo de la misma forma en que lo hacen los estudios con el genoma completo. Con este fin se propone una metodología que parte del análisis descriptivo de la variabilidad de las subregiones del genoma, acompañado de un análisis estadístico, continúa con la evaluación de la señal filogenética a partir de la saturación y el ruido, y termina evaluando la capacidad de cada región de reconstruir filogenias o redes equivalentes a las obtenidas con la información del genoma completo.
MATERIALES Y MÉTODOS
Para el presente estudio se tuvieron en cuenta las características de las distintas subregiones del genoma del SARS-Cov-2 expuestos por Wu, Zhao y colaboradores (Wu, Zhao et al., 2020). El genoma de aproximadamente 29,903 nucleótidos (nt) se organiza de forma similar a los virus del género Betacoronavirus, siguiendo a dirección 5 'a 3', se define su composición génica con la replicasa ORF1ab(21,291 nt, su producto es una poliproteína que tiene como productos de escisión predichos 16 proteínas no estructurales), S (que codifica la glucoproteína de espiga estructural), ORF3a (proteína de ORF3a), E (proteína estructural de la envoltura), M (glucoproteína estructural de membrana), ORF6 (proteína ORF6), ORF7a (proteína ORF7a), ORF7b (proteína ORF7b), ORF8 (proteína ORF8), ORF9 (incluye N, fosfoproteína estructural de la nucleocápside) y ORF10 (proteína ORF10). Adicionalmente, tiene secuencias terminales 5 'y 3' que son típicas de los betacoronavirus, con 265 nt en el extremo 5' terminal y 229 nt en el extremo terminal 3'.
Con el objetivo de evaluar su potencial uso como marcadores filogenéticos, se siguió una metodología que se puede resumir en los siguientes pasos:
Selección de las subregiones de mayor variabilidad a partir de la presencia de varias posiciones polimórficas y la existencia de diferencias entre áreas geográficas en los patrones de variabilidad en dichas subregiones.
Evaluación de la señal filogenética en las subregiones seleccionadas, en comparación con la señal filogenética de todo el genoma, teniendo en cuenta la existencia de saturación y ruido.
Evaluación de la capacidad relativa de las subregiones seleccionadas para reproducir las relaciones evolutivas, con respecto a las construidas con la información de todo el genoma a partir de comparaciones topológicas y probabilísticas de las topologías obtenidas con la subregión y con todo el genoma.
El estudio inicial de la variabilidad por regiones se realizó mediante la herramienta para el análisis de variabilidad disponible en la plataforma Nextstrain (https://nextstrain.org/). Se realizó un filtrado inicial de las regiones del genoma, partiendo del criterio que un buen marcador filogenético debe ser suficientemente variable como para ser informativo.
Para el estudio de la influencia de la región geográfica en la variabilidad, se realizó la comparación por la prueba estadística de Friedman (Friedman, 1940) seguida de la comparación por pares usando Wilcoxon (Wilcoxon, 1992) corregido por el criterio de Holm (Holm, 1979) para comparaciones múltiples, de la entropía por sitio de todo el genoma entre el comportamiento global y el de seis áreas geográficas: Asia, Europa, América del Norte, América del Sur, África y Oceanía.
El mismo análisis se realizó para cada región del genoma en particular, con el objetivo de evaluar si el patrón de comportamiento es el mismo que a nivel de genoma. Estos análisis fueron realizados en el paquete estadístico R (Core, 2017). Se incluye una evaluación de la asociación existente entre las áreas geográficas y las diferentes clasificaciones taxonómicas de las secuencias, para lo cual se evaluó la capacidad de las clasificaciones de Nextstrain y GISAID, tomadas como variables independientes, de predecir el área geográfica de origen, mediante una derivación del método CHAID (Biggs, De Ville et al., 1991), el CHAID exhaustivo, implementado en el programa IBM SPSS v20 (IBM). Estos procesamientos se realizaron empleando los metadatos disponibles para su descarga en la plataforma Nexstrain.
Para los estudios filogenéticos se trabajó con las mismas secuencias empleadas en (Forster, Forster et al., 2020), cuya tabla de agradecimientos (con los códigos de acceso a GISAID, y datos de origen y autores) aparece en el Material Suplementario D. Se empleó este conjunto de datos porque permite evaluar la capacidad de la inferencia filogenética para reconstruir rutas de infección y se toman casos de estudio para los que existía un historial de viajes conocido.
Los genomas completos se alinearon utilizando la herramienta progressiveMauve (Darling, Mau et al. 2010). Se tomaron como potenciales marcadores las regiones codificantes del genoma del SARS-Cov-2. Las diferentes regiones se obtuvieron de las secuencias genómicas utilizando la anotación asociada al ensamblaje GCF_009858895.2 del NCBI, tomando como referencia la secuencia con código NC_045512.2 del GenBank correspondiente al aislado Wuhan-Hu-1, mediante el programa BLAT v 36x5 (Kent, 2002). Los conjuntos de secuencias para cada región se alinearon con el programa MAFFT v7 (Katoh and Standley, 2013), empleando el marco de trabajo de herramientas de análisis bioinformático del EMBL-EBI (Goujon, McWilliam et al., 2010). En todos los casos los alineamientos se procesaron con el software Gblocks (Castresana, 2000) versión 9b, para eliminar las regiones pobremente alineadas que pudieran afectar la calidad de los análisis.
Los efectos de saturación se investigaron trazando el número absoluto de transiciones y transversiones versus distancia genética para todos los CV-A24v, utilizando el software DAMBE v7.2.43 (Xia and Xie, 2001) y la prueba de Xia et al. 2003 (Xia, Xie et al., 2003) disponible en el mismo software, en ambos casos usando el modelo evolutivo de Felsenstein (F84) (Felsenstein and Churchill, 1996) para el cálculo de las distancias genéticas. Se realizó el estudio en las posiciones 1 + 2 + 3, teniendo en cuenta que, de aparecer saturación, se evaluarían las posiciones 1+2 y 3. El ruido en la señal se evaluó mediante el método de mapeo de verosimilitud (Strimmer and von Haeseler, 1996), implementado en el programa Tree-Puzzle v 5.3 (Schmidt, Strimmer et al., 2002). Antes de cada procesamiento, las secuencias idénticas fueron eliminadas del conjunto de datos, empleando el propio programa DAMBE v7.2.43.
Los estudios filogenéticos preliminares se realizaron utilizando el método de máxima verosimilitud (ML) mediante el programa IQ-TREE (Nguyen, Schmidt et al., 2015), mediante el servidor web W-IQ-TREE (Trifinopoulos, Nguyen et al., 2016) disponible en http://iqtree.cibiv.univie.ac.at/. Para cada conjunto de taxones se realizó la reconstrucción filogenética para el genoma completo y para cada región genómica. Posteriormente se emplearon los métodos KS (Kishino, Miyata et al., 1990), SH (Shimodaira and Hasegawa, 1999) y AU (Shimodaira, 2002) para comparar el soporte de las topologías obtenidas por cada región respecto a la obtenida con todo el genoma, utilizando 10000 réplicas. Los árboles se visualizaron y editaron empleando el programa FigTree v 1.4.3 (Rambaut, 2017).
Adicionalmente se realizó la comparación de las topologías obtenidas mediante el programa ETE Toolkit versión 3 (Huerta-Cepas, Serra et al., 2016), para determinar el nivel de similitud entre las topologías obtenidas por regiones y la obtenida con todo el genoma. Se compararon los árboles máximo verosímiles obtenidos por IQ-TREE, empleando la métrica RF (Robinson and Foulds, 1981).
RESULTADOS Y DISCUSIÓN
El primer paso para la selección de los posibles marcadores filogenéticos en la secuencia de SARS-Cov-2 consistió en el análisis visual de la variabilidad por regiones. En el Material Suplementario A se muestran los gráficos ampliados para la entropía de todas las subregiones. Se observa que, si bien los eventos de mutaciones comprenden toda la extensión del genoma, en general se caracteriza por pocos eventos de mutación (pocos eventos y baja entropía) en la mayoría de las regiones, y solo hay algunos puntos calientes (hot spots), en las regiones del ORF1 (a y b), en la región que codifica a la espiga (S), el ORF3a, ORF8, el gen N de la fosfoproteína de la nucleocápside (ORF9), y el ORF14. El ORF14, al no estar caracterizado, no está incluido en la tabla de anotación del genoma empleada para el estudio, lo cual dificultaba el procesamiento automático de las secuencias, por lo que no fue considerado para el estudio.
Esta situación también se ve matizada por la relativamente baja tasa de mutación del SARS-Cov-2 (para un virus de ARN). Entre las herramientas que brinda la plataforma Nextstrain es el estimado de la tasa de variación del virus, que hasta la fecha se estima en 24.884 (esta tasa se actualiza por días) mutaciones por año (ver Fig. S 14). La falta de diversidad puede limitar el poder de resolución de los métodos filogenéticos para asignar un origen ancestral, al reducir la cantidad de sitios filogenéticamente informativos, pero al mismo tiempo hace más relevantes las mutaciones específicas que puedan indicar una ruta evolutiva determinada. Como la diversidad genética del SARS-CoV-2 es baja durante el período pandémico temprano, se esperaría una asociación directa entre la asignación de linaje y la presencia de conjuntos particulares de mutaciones (Rambaut, Holmes et al., 2020). El comportamiento en la variabilidad de las secuencias de SARS-Cov-2 en el curso actual de la pandemia, correspondiente a la fase de expansión, implica unas condiciones cercanas a las estipuladas por el modelo de mutaciones infinitas de Kimura (Kimura, 1969), en el cual se asume que hay un número infinito de sitios donde pueden ocurrir mutaciones y cada nueva mutación ocurre en un sitio nuevo. La tasa estimada de mutaciones por año en conjunto con la longitud del genoma, cercana a las 30 Kb, hace poco probables las mutaciones en la misma posición o en posiciones cercanas del genoma.
Para determinar si existen diferencias significativas en los patrones de variabilidad entre las diferentes áreas geográficas, tanto a nivel de genoma como a nivel de segmentos, se realizaron las comparaciones estadísticas correspondientes. En el Material Suplementario B se muestran las salidas de las pruebas estadísticas. En todos los casos la prueba de Friedman arroja la existencia de diferencias estadísticamente significativas globales en las entropías por posiciones de secuencias. En las comparaciones por pares, cuando se emplea la información de todo el genoma encuentran diferencias estadísticamente significativas entre todas las áreas geográficas, salvo entre Sudamérica y Oceanía (por submuestreo, no por diferencias reales), y entre todas estas y el valor global. En las comparaciones por subregiones del genoma se puede ver, entre los resultados más notables, que en el ORF1a muestra un comportamiento similar al del genoma completo, no así el resto de las regiones.
Al evaluar la capacidad predictiva de las clasificaciones Nextsatrain y GISAID de las variantes de SARS-Cov-2, como variables predictivas del área geográfica, se puede observar (Figura 1) que, en primer lugar, el sistema de clasificación ofrecido por Nextstrain es el más infuyente en la predicción de del área geográfica (es la única variable que aparece en el árbol resultante). Incluso esta variable es un mal predictor del área geográfica, pues solo clasifica las secuencias en dos categorías: Asia y África.
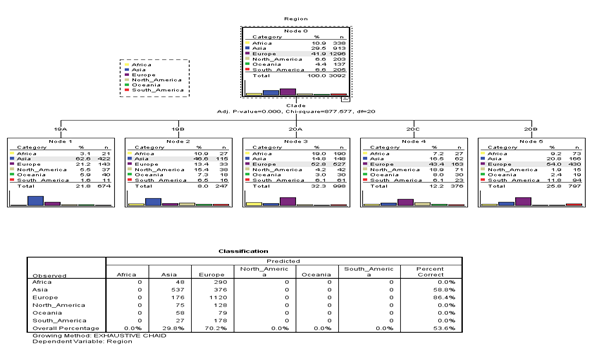
Fig. 1 Resultados de procedimiento de clasificación mediante árboles por el algoritmo CHAID, realizado en el SPSS v 20. Como variables predictivas se emplearon las clasificaciones Nextstrain (la única reflejada en el árbol al ser la más influyente) y la clasificación GISAID. Se emplearon los metadatos de 3092 secuencias muestreadas en GISAID.
En el preprocesamiento de los alineamientos usando el Gblocks y Dambe para eliminar regiones mal resuetas y secuencias repetidas, se puede apreciar una notable reducción de la cantidad de secuencias no idénticas en la medida en la que se reduce la longitud de la región del genoma analizada. Los datos empleados en el trabajo de (Forster, Forster et al., 2020), además de incluir una secuencia de coronavirus de murciéago, RaTG13, muestreada en el 2013, cubren solo el período de diciembre de 2019 a febrero de 2020, cuando el virus apenas se estaba expandiendo por el mundo, y se había acumulado muy poca diversidad. Estas características afectan, por supuesto, cualquier generalización que se presente basada en este conjunto de datos. En lo que sigue, salvo para la evaluación de la saturación, se mantuvieron todas las secuencias para observar la influencia real del muestreo taxonómico en los resultados. Para constrastar algunos resultados, se repitieron con un conjunto de 51 secuencias provenientes de Bélgica (Material Suplementario H).
En el Material Suplementario C se muestran los resultados del análisis de saturación realizado con Dambe, así como de la evaluación de ruido por mapeo de verosimilitud realizado en Tree-Puzzle, en el conjunto de datos correspondiente al trabajo de (Forster, Forster et al., 2020). Como era de esperarse, no se observa saturación, y el test de Xia (Xia, Xie et al., 2003) lo confirma en todos los casos (solo fue necesario evaluar el esquema 1+2+3).
El ruido, por su parte, tiene un comportamiento notablemente deficiente. Como se anunciaba anteriormente, con tantas secuencias similares o idénticas, es de esperar que la resolución de las filogenias no sea elevada. Según se describe en (Schmidt and von Haeseler, 2009), desde un punto de vista biológico, un análisis de mapeo de verosimilitud que muestra más del 20% -30% de los puntos en el área correspondiente a topologías de tipo estrella o red sugiere que los datos no son confiables para la inferencia filogenética (al menos para la obtención de árboles). En el Material Suplementario C se puede observar que incluso para el genoma completo, el 50.9 % de los puntos cumplen con este criterio (el 48.3 % lo hace en la región totalmente no resuelta, tipo estrella), lo cual hace muy poco resolutivas la inferencia filogenética, lo cual es un factor de peso a tener en cuenta. En el Material Suplementario H (Figura.S. 15) se puede observar el resultado del mismo análisis para el conjunto de datos proveniente de Bélgica. Se puede observar que, incluso en este caso, la capacidad resolutiva se ve afectada, con un 28.5 % de los puntos en las regiones de no resolución, y predominio en la región central con un 27.4 %.
En la Fig.2 se observa la salida con los valores de significación para las pruebas KH, SH y AU resultantes de comparar las filogenias obtenidas mediante filogenómica y las obtenidas con cada una de las regiones. Se puede observar que de acuerdo a las pruebas KH y AU, solo el ORF1a permite obtener árboles cuya verosimilitud indica que son igualmente probables que el árbol obtenido con el genoma completo. Para la prueba SH ocurre lo mismo con el ORF3a y el ORF8, pero hay que tener en cuenta que esta prueba tiende a ser muy conservadora (Shimodaira, 2002), por lo tanto, el resultado más confiable es el aportado por la prueba AU. Los segmentos M y ORF6 no tienen sitios parsimoniosamente informativos, por lo que no se pudo realizar la comparación con ellos. En el Material Suplementario D (Figura 2) se puede observar que, en los datos provenientes de Bélgica, además del ORF1a, también el árbol construido con la subregión de la espiga muestra un soporte comparable al del genoma completo. En este caso solo se consideraron las subregiones genómicas ORF1a, S, ORF3a, ORF8 y N, para mostrar el comportamiento en segmentos de varias longitudes.

Fig. 2 Salida del IQ-TREE para el procedimiento de comparación de topologías, realizado con la información del genoma completo y los respectivos árboles construidos con las subregiones genómicas, partiendo de las secuencias empleadas en el trabajo de (Forster, Forster et al. 2020).
La comparación de topologías se realizó con ETE Tolkit, comparando las topologías de los árboles máximo verosímiles obtenidos en IQ-TREE. La Figura 3 muestra la salida del procedimiento ete-compare. En la figura:
nRF: distancia normalizada de Robinson-Foulds (RF / maxRF)
RF: Distancia simétrica de Robinson-Foulds
maxRF: valor máximo de Robinson-Foulds para esta comparación
% src_br: frecuencia de ramas en el árbol de destino encontrado en el árbol de referencia (1.00 → se encuentra el 100% de las ramas)
% ref_br: frecuencia de ramas en el árbol de referencia encontrado en el árbol de destino (1.00 → se encuentra el 100% de las ramas)
Número de subárboles utilizados para la comparación (se aplica solo cuando se utilizan elementos duplicados para descomponer árboles objetivo)
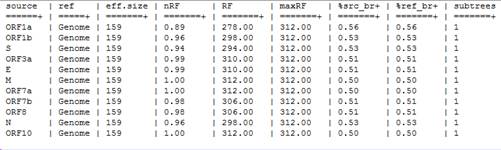
Fig. 3 Comparación entre las topologías de los árboles obtenidos con subregiones genómicas con respecto al obtenido con el genoma completo, realizada con el software ETE Tolkit.
Se puede observar, en concordancia con lo analizado anteriormente, que la topología menos distante (más similar) a la obtenida con el genoma completo, se obtiene con el ORF1a. No obstante, es notable que solo comparten el 56% de las ramas, por lo que existen diferencias considerables entre las topologías. Este valor no es muy diferente del obtenido con las otras subregiones. El segmento S le sigue en similitud a la topología del árbol construido con el genoma completo, y le siguen N y ORF1b. En el Material Suplementario D (Figura 4) se observa que con los datos de Bélgica ocurre algo similar. La Figura.S. 18 muestra las topologías de los árboles máximo verosímiles obtenidos con IQ-TREE para las secuencias belgas. Se observa que en la inferencia realizada a nivel de genoma el árbol está mucho más resuelto que los que se obtienen en la medida en que se asciende en el valor de distancia dado por RF, según se reporta en la tabla de la ¡Error! No se encuentra el origen de la referencia.. En general se puede ver que para la reconstrucción de filogenias partiendo de datos de secuencias del SARS-Cov-2 en su estado actual de evolución (para el momento de realizarse este estudio), tanto en lo referido a la topología como al soporte, la fortaleza de la señal filogenética, y por lo tanto la probabilidad de reconstruir el historial evolutivo de las secuencias; está estrechamente ligada a la longitud del segmento seleccionado.
En la Fig. 4 se muestran los árboles máximo verosímiles obtenidos en IQ-TREE para los mismos datos, realizados empleando todo el genoma, el ORF1a y el segmento N. Sobre la capacidad de la red filogenética para reconstruir rutas de infección donde son desconocidas y representan un riesgo para la salud pública, en el trabajo de (Forster, Forster et al. 2020) se toman, entre otros, como casos de estudio, los primeros aislamientos realizados en Brasil (Brazil/SPBR-02/2020|EPI_ISL_413016|2020-02-28) y en México (Mexico/CDMX-InDRE_01/2020|EPI_ISL_412972|2020-02-27, (Garcés-Ayala, Araiza-Rodríguez et al., 2020)). Para ambos casos existía un historial de viajes conocidos, y en el caso de la secuencia mexicana (Forster, Forster et al., 2020) afirman que se puede trazar la cadena de contagios a partir de información epidemiológica conocida hasta el foco de infección en Wuhan. Se observa la dependencia con respecto a la fortaleza de la señal (hay baja resolución de las relaciones filogenéticas) y a un adecuado muestreo de taxones (que en definitiva es lo que permite, aún en el marcador menos informativo, el de la subregión N, todavía poder inferir un origen geográfico de la secuencia mexicana). En las figuras ¡Error! No se encuentra el origen de la referencia. y ¡Error! No se encuentra el origen de la referencia. se muestra la inferencia filogeográfica realizada en la plataforma Nexstrain, empleando la reconstrucción de estados en el contexto de la inferencia por máxima verosimilitud, y empleando un muestreo voluminoso de las secuencias existentes en GISAID. Para la secuencia brasileña, que pertenece al subtipo 20A de Nextstrain (y al V de GISAID) se infiere un origen europeo, aunque no italiano, como lo indica el conocimiento epidemiológico previo. Lo mismo ocurre con la secuencia mexicana (20B de acuerdo a Nextstrain, GR de acuerdo a GISAID). Este resultado muestra, que incluso con un muestreo taxonómico mayor, si no existe específicamente un muestreo en posiciones cercanas a la ruta de transmisión, la precisión de la inferencia se reduce.
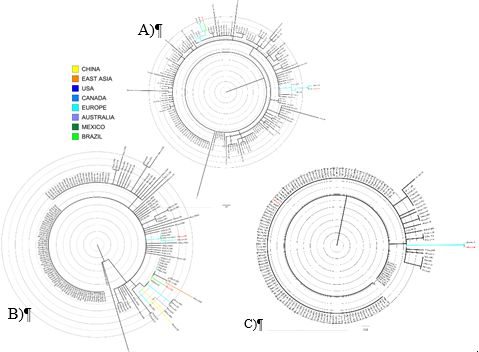
Fig. 4 Árboles máximo verosímiles obtenidos en IQ-TREE para el conjunto de secuencias empleado en (Forster, Forster et al., 2020) a partir de A) todo el genoma, B) la subregión ORF1a y C) la subregión N.
Este estudio está limitado por los conjuntos de datos empleados, aunque algunas de las conclusiones surgen del análisis de los datos reflejados en la plataforma Nextstrain, donde se ofrece información basada en un muestreo bastante amplio de las cepas virales circulantes por todo el mundo. El análisis se basó en regiones del genoma delimitadas por la función, así que se analizaron los marcos abiertos de lectura en su totalidad y por separado, pero nada obliga a que un marcador filogenético esté restringido a un único marco abierto de lectura. Se deben realizar estudios posteriores que permitan, con un procedimiento automatizado y criterios adecuados de optimalidad, la selección específica de un segmento del genoma.
CONCLUSIONES
La evidencia que se presenta en este trabajo, permite resaltar algunos aspectos de la evolución presente del SARS-Cov-2, con relevancia para la selección de marcadores filogenéticos. Uno de los aspectos fundamentales es que el relativamente bajo nivel de diversidad que presenta todavía el genoma viral, deriva en una señal filogenética débil incluso a nivel de genomas, pero que pudiera mostrar un notable declive en la medida en la que se analicen fragmentos más pequeños.
Este comportamiento sugiere que, en el estado de evolución del virus en el momento en que fue realizado el estudio, los aspectos aleatorios, asociados a la evolución neutral, pudieran estar teniendo mayor influencia en los resultados de la inferencia filogenética que los aspectos asociados a presiones selectivas. La capacidad resolutiva de los métodos de reconstrucción filogenética está limitada por la baja fortaleza de la señal filogenética, y el adecuado muestreo taxonómico determinará el valor de la inferencia filogenética para cada estudio en particular. No parece existir, en concordancia con la baja diversidad observada, un número elevado de homoplasias que interfieran en la correcta inferencia de las relaciones evolutivas usando árboles.
De acuerdo a lo que refleja el trabajo, el ORF1a sería el marcador cuyo uso reproduciría de forma más fiable las relaciones evolutivas que se pueden inferir a nivel de genomas, pero todo apunta a que se debe a su longitud (cerca de 14 kb), lo cual no los haría factible para su obtención por el método Sanger usando solo un par de cebadores. Las regiones cuya longitud están en el orden del largo de lectura en el método Sanger (alrededor de 900 pb) no muestran un buen comportamiento como marcadores filogenéticos.

