










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
Dentro de la familia Trypanosomatidae hay dos géneros de importancia médica por su papel como patógenos humanos, Trypanosoma y Leishmania. Dentro de estos géneros se encuentran 21 especies de Leishmania, Trypanosoma cruzi y subespecies de Trypanosoma brucei (T. brucei gambiensi y T. brucei rhodesiense), agentes causantes de la leishmaniasis, la Tripanosomiasis Americana (Enfermedad de Chagas) y la Tripanosomiasis Africana Humana (Enfermedad del sueño), respectivamente. En conjunto, estas infecciones afectan a más de 30 millones de personas en todo el mundo y representan un grave problema de salud pública, especialmente en África, América del Sur y Asia. Estas enfermedades son responsables de altas tasas de mortalidad y morbilidad en los países en desarrollo y tienen un impacto económico y social en las regiones afectadas. Entre los desafíos para el control de estas enfermedades se encuentra contar con métodos de diagnósticos sensibles y específicos más fáciles de utilizar, especialmente en áreas remotas donde estas enfermedades son endémicas y que permitan la detección, identificación, diferenciación y caracterización de casos a mejor costo-efectividad. Sin embargo, para diseñar una herramienta diagnóstica fiable es importante llegar a un consenso sobre la clasificación taxonómica de los géneros Leishmania y Trypanosoma, ya que la validez del esquema de clasificación taxonómica de estos todavía se debate y el estado de algunas especies se cuestiona.
La clasificación taxonómica del género Leishmania se basa principalmente en criterios ecobiológicos como el vector, la distribución geográfica, el tropismo, las propiedades antigénicas y las manifestaciones clínicas (1. Se han propuesto esquemas taxonómicos jerárquicos utilizando categorías de subgéneros, complejos de especies, especies y subespecies. Los dos subgéneros (L.) Leishmania y (L.) Viannia se separan en función de su ubicación en el intestino del vector 1, y las especies dentro de los subgéneros generalmente se establecieron mediante Electroforesis de Enzimas Multilocus (MLEE), 1 hasta la fecha la regla de oro. La validez del esquema de clasificación taxonomíca ha sido cuestionado varias veces, y el debate se centra en el estado de algunas especies como: L. (V.) panamensis, L. (V.) peruviana, L. (L.) infantum, L. (L.) chagasi, L. (L.) archibaldi, L. (L.) garnhami, L. (L.) pifanoi y L. (V.) lainsoni.3 Además, la posición taxonómica y la relación filogenética de Leishmania de lagartos sigue siendo incierta. (1 Por lo tanto, es necesario acordar un esquema práctico de clasificación taxonómica basado en conceptos confiables y consensuales. Una buena definición de las especies de Leishmania es crucial para el diagnóstico correcto y el pronóstico de la enfermedad, así como para tomar decisiones sobre el tratamiento y las medidas de control. 4
La clasificación taxonómica del género Trypanosoma se basa en la establecida por Hoare quien la basa en la morfología y el desarrollo del vector. (5 Las especies de Trypanosoma que infectan mamíferos se separan en dos secciones: Salivaria y Stercoraria. Los Trypanosomas salivares se subdividieron aún más en cuatro subgéneros: Trypanozoon, Duttonella, Nannomonas y Pycnomonas. La sección Stercoraria comprende tres subgéneros: Schizotrypanum, Megatrypanum y Herpetosoma.6 Dado que algunos de estos grupos son polifiléticos y carecen de relevancia evolutiva y taxonómica, recientemente, algunos autores sugirieron clasificar el género Trypanosoma en clados filogéneticos basado en la subunidad pequeña ARNr (SSU-rRNA) y gliceraldehido 3´fosfato deshidrogenasa (gGAPDH).7-10
Los estudios filogenéticos moleculares han sido y sigue siendo una técnica que dominan los estudios taxonómicos modernos. Recientemente se ha propuesto un nuevo sistema de clasificación de eucariotas basados en datos de enfoques morfológicos modernos, vías bioquímicas y filogenia molecular.
Diferentes marcadores moleculares se han utilizado, dentro de ellos el gen que codifica la proteína de choque térmico 70 (HSP70), el que se ha utilizado como un marcador evolutivo importante en diferentes estudios de parásitos (Cryptosporidium spp. Babesia spp., Giardia spp., Entamoeba spp., Microsporidium spp. y Blastocystis hominis). Los genes que codifican la HSP70 citoplasmática se encuentran entre los primeros genes de Kinetoplastideos en ser clonados y caracterizados debido a su naturaleza conservada. Esta proteína es altamente conservada en secuencia y función, tiene una gran importancia como chaperona molecular, en el plegado de la proteína y como transporte. El gen se organiza en repeticiones en tándem de la cabeza a la cola y el número de copia depende de la especie. 11
En este estudio investigamos las relaciones filogenéticas de los géneros Leishmania y Trypanosoma utilizando la secuencia del gen hsp70 citoplasmática y discutimos las implicaciones para las definiciones de especies, la evolución y la taxonomía de estos géneros.
Métodos
Cepas o aislamientos de especies de Leishmania y Trypanosoma utilizados en el estudio
Se utilizaron en el estudio ADN genómico procedente de 44 cepas o aislamientos de 11 especies de Leishmania y 41 cepas o aislamientos de 9 especies de Trypanosoma de diferentes orígenes geográficos, donados por diferentes laboratorios y que forman parte de la colección de la Unidad de Parasitología Molecular del Instituto de Medicina Tropical de Amberes, Bélgica.
PCR-hsp70
Se amplificó un producto de PCR de 1422 pb de las cepas o asilamientos estudiados utilizando el juego de cebadores descritos por Garcia et al. 12 La mezcla de reacción (50 ml) contenía 1x de tampón de PCR que incluye 1,5 mM MgCl2, 1x tampón Q, 200 µM de cada desoxinucleósido trifosfato, 0.5 U HotStarTaq Plus DNA polimerasa (Qiagen, Hilden, Alemania), 0,4 mM de cada cebador, y alrededor de 10 ng de ADN genómico aislado del cultivo de parásitos. Las condiciones en el termociclador fueron: desnaturalización inicial 95 °C por 5 min; seguido de 35 ciclos que consisten en 94 °C durante 40 s, 61 °C por 1 min, 72 °C por 2 min; y un paso final de extensión de 8 min a 72 °C. Los amplicones se analizaron en un gel de agarosa al 2 % y fueron secuenciados directamente sin clonación molecular.
Secuenciación del gen hsp70
Se realizó la secuenciación de los productos de amplificación de la PCR-hsp70 de las cepas o aislamientos de especies de Leishmania y Trypanosoma. A partir de las secuencias de 8 cebadores diseñados para este propósito, 13-14 se secuenciaron ambas cadenas, excepto los 40 nucleótidos contenidos en los extremos correspondientes a la región complementaria a los cebadores de los que se secuencio una cadena solamente. Para obtener la secuencia se utilizó el juego ¨BigDye ™ Terminator cycle-sequencing-ready-reaction kit¨ (Perkin Elmer, Foster City, CA, EUA) y el secuenciador automático ABI 3730 (Perkin Elmer). Para el análisis filogenético, solo la región entre los cebadores fue retenida, es decir, 1380 de los 1422 nucleótidos. Se obtuvo que la secuencia consenso de cada cepa estudiada utilizando el programa Clustal X 15 con ajuste manual. Las secuencias del gen hsp70 adicionales se obtuvieron de la búsqueda en BLAST en las siguientes bases de datos: GeneDB (http://www.genedb.org), TritrypDB (http://www.tritrypdb.org) y GenBank (http://www.ncbi.nlm.nih.gov).
Análisis filogenético
Las secuencias obtenidas se alinearon con las secuencias publicadas previamente en las bases de datos (10 cepas o aislamientos de Leishmania spp y 24 cepas o aislamientos de Trypanosoma spp), utilizando el paquete de software MEGA (Molecular Evolutionary Genetic Analysis Version 5.05, 16 www.megasoftware.net).
Las relaciones filogenéticas entre las distintas especies estudiadas, se analizaron en base a la información obtenida de las secuencias del gen hsp70, en comparación con dos cepas de Trypanosoma que se utilizaron como grupos externos para el análisis filogenético del género Leishmania y 14 especies de Leishmania y Paratrypanosoma confusum para el análisis del género Trypanosoma.
El programa MEGA se utilizó para la construcción de distintos arboles basados en parámetros de distancia (método de unión al vecino, “neighborg joining”, utilizando el modelo Kimura-2 parameter, 17 y caracteres (máxima parsimonia, “maximun parsimony” 18-19 y máxima verosimilitud, maximun likelihood” 20, este último bajo el modelo “general time reversible and gamma distributed”). Las distancias de las secuencias aminoacídicas fueron determinadas mediante el modelo “p-distance”. El soporte de los grupos monofiléticos se determinó por el método de boostrap.20
Utilizando las mismas secuencias nucleotídicas se construyó una red filogenética utilizando el método de “Neighbor-Net”, modelo “p-distance” no corregido utilizando el programa SplitsTree4. 21-22
Resultados y discusión
Secuenciación y análisis del polimorfismo genético de los géneros Leishmania y Trypanosoma
Se obtuvo la secuencia de nucleótidos de un fragmento de 1380pb del gen hsp70 citoplasmático de 44 cepas y aislamientos de diferentes orígenes geográficos que pertenecen a 16 especies de Leishmania. Reportándose por primera vez para la secuencia de este gen para 11 especies de Leishmania: L. (V.) peruviana, L. (V.) guyanensis, L. (V.) panamensis, L. (V.) naiffi, L. (V.) lainsoni, L. (L.) mexicana, L. (L.) garhnami, L. (L.) tropica, L. (L.) aethiopica, L. (L.) chagasi y L. (L.) archibaldi; para el resto de las especies se reportaron secuencias adicionales. Así mismo se obtuvo la secuencia de nucleótidos de 41 cepas y aislamientos de diferentes orígenes geográficos para 9 especies de Trypanosoma: T. cruzi, T. rangeli, T. brucei, T. congolense, T. evansi, T. vivax, T. equiperdum, T. theileri y T. lewisi. Reportándose por primera vez para las Unidades Discretas de Tipificación (DTU) de T. cruzi TcIII, TcIV y TcV, además de T. brucei gambiense, T. brucei rhodesiensi, T. equiperdum, T. theileri y T, lewisi. Igualmente, para el resto de las especies, se reportaron secuencias adicionales. Para ambos géneros las secuencias obtenidas se depositaron en el Banco de Genes (GeneBank). Para las cepas correspondientes a las diferentes especies de Leishmania los números de acceso son: FN395020-FN395056 y EU599090-EU599093, mientras que para las cepas correspondientes a las especies de Trypanosoma los números de acceso son: KC959988-KC960011, KP208734-KP208748 y KP257564-KP257565.13 - 14
Para el género Leishmania, el fragmento del gen hsp70 es rico en guanina-citosina (GC) (63,6-65,4 %), con una similitud entre las secuencias analizadas entre 94,1 y 99,8 %. La variación de la secuencia nucleótidos es suficiente para discriminar especies de parásitos: 148 posiciones nucleotídicas polimórficas (11 %) y 111 posiciones nucleotídicas parsimoniosamente informativas (8 %), lo que significa que al menos dos nucleótidos diferentes están presentes, cada uno encontrado en al menos dos secuencias. Las secuencias aminoacídicas deducidas de las secuencias nucleotídicas (459 aminoácidos) revelaron sustituciones en 43 posiciones (9 %), de los cuales 39 sitios (8 %) son parsimoniosamente informativos. 13
Para el género Trypanosoma, el fragmento del gen hsp70 es igualmente rico en guanina-citosina (GC) (de 55,8 % a 65,6 %), con una similitud entre las secuencias analizadas entre 75 % y 100 %. La variación de la secuencia nucleótidos es suficiente para discriminar especies de parásitos: 526 posiciones nucleotídicas polimórficas (29,8 %) y 412 posiciones nucleotídicas parsimoniosamente informativas (29,8 %). Las secuencias aminoacidicas deducidas de las secuencias nucleotídicas (459 aminoácidos) revelaron sustituciones en 132 posiciones (28,8 %), de los cuales 83 sitios (18,1 %) son parsimoniosamente informativos.14
La variación identificada entre las especies de Leishmania y entre las especies de Trypanosoma apoyadas en la existencia de sitios parsimoniosamente informativos, habla de que el gen es un marcador molecular apropiado para la tipificación de especies, subespecies y linajes de ambos géneros. 13-14
Al analizar la relación entre el número de sustituciones no sinónimas por sitios potencialmente no sinónimos (dN) y el de sustituciones sinónimas por sitios potencialmente sinónimos (dS), para ambos géneros resulto la relación dN/dS<1 (Leishmania dN/dS = 0,30; Trypanosoma dN/dS = 0,08), lo que representa que este gen para ambos géneros está sujeto a una selección purificadora que ha permitido preservar la funcionalidad de la HSP70. 13-14
Análisis filogenético del género Leishmania
La Figura 1 muestra el árbol filogenético construido a partir de las secuencias nucleotídicas utilizando el método de Unión al Vecino. Los subgéneros L. (Leishmania) y L. (Viannia) forman un grupo monofilético, con L. (Sauroleishmania) ramificándose en el medio como un taxón independiente. Dentro del subgénero L. (Leishmania), el árbol muestra una distinción entre especies del Viejo y Nuevo Mundo. 13
El análisis filogenético soporta ocho grupos monofiléticos: L. (L.) donovani (Complejo L. (L.) donovani), L. (L.) major, L. (L.) tropica (Complejos L. (L.) aethiopica y L. (L.) tropica), L. (L.) mexicana (Complejo L.(L.) mexicana), L. (V.) lainsoni, L. (V.) naiffi, L. (V.) guyanensis (Complejo L. (V.). guyanensis y L. (V.) braziliensis (Complejo L. (V.). braziliensis). Estos grupos fueron también verificados en árboles filogenéticos construidos por los métodos de máxima parsimonia y máxima verosimilitud, así como los árboles obtenidos basados en las secuencias aminoacídicas deducidas (datos no mostrados). 13
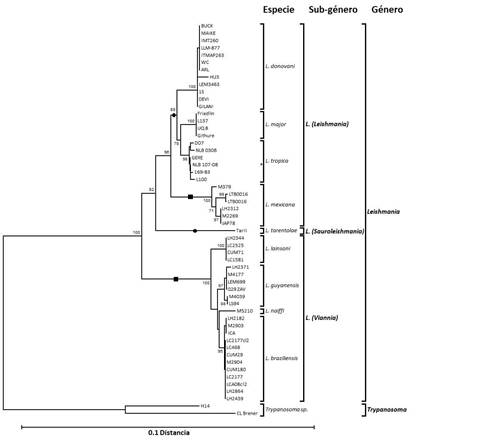
Fig. 1 Árbol filogenético basado en las secuencias del gen hsp70 de distintas especies de Leishmania según el método de unión al vecino (neighbor-joining). La distancia fue estimada por el método de Kimura 2-parameter. El análisis de “bootstrap” 2000 réplicas. Especies del viejo Mundo se representan con círculos en la rama, mientras que especies del Nuevo Mundo con un cuadrado. El árbol está enrutado con las especies de Trypanosoma. 13
Como se puede observar en la figura 2A, los mismos grupos que se identificaron por el análisis convencional se muestran en esta red. Una ampliación del grupo que comprende las especies del subgénero L. Viannia (figura 2B) muestran que L. (V.) panamensis y L. (V.) peruviana se encuentran como subgrupos separados dentro de los complejos L. (V.) guyanensis y L. (V.). braziliensis, respectivamente. De la misma forma que se muestra como L. (L.). infantum, aparece como subgrupo separado dentro del complejo L. (L.) donovani (figura 2C). De ahí que concluyamos que las especies L. (L.) infantum, L. (V.) panamensis y L. (V.) peruviana se encuentran como subgrupos separados dentro de los complejos L. donovani, L. guyanensis y L. braziliensis respectivamente. Las especies L. (L.) aethiopica, L. (L.) amazonensis y L. (L.) garnhami no pueden distinguirse como entidades separadas (figura 2 C, D). 13
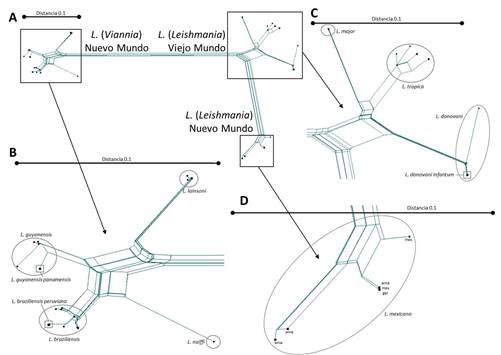
Fig. 2 Red filogenética basada en las secuencias del gen hsp70 de distintas especies de Leishmania, según el método “NeighborNet algorithm” cada uno de los cuatro paneles (A)- (D) se dibuja a la escala indicada. (A). Red completa con representaciones de los tres grupos mostrados con más detalles en los paneles restantes. (B.) Las secuencias del subgénero L. (Viannia) se separan en cuatro especies. Los cuadrados se usan para indicar subespecies. (C). Secuencias del subgénero L. (Leishmania) del Viejo Mundo, separados en tres especies. Dentro de L. (L.). Tropica, los cuadrados (tro) denotan las secuencias de L. (L.) tropica, los puntos (aet) representan L. (L.) aethiopica. Dentro de L. (L. donovani), los cuadrados representan L. donovani infantum. (D). Secuencias del Nuevo Mundo del subgénero L. (Leishmania). Las especies L. (L.) garnhami, L. (L.) mexicana y L. (L.) amazonensis están indicadas por gar, mex y ama, respectivamente. Figura de artículo.13
Con vistas a corroborar los resultados obtenidos basados en el gen que codifica la HSP70 se realizó el análisis combinado de hsp70+proteína de choque termino pequeña 20Kda (hsp20). Las secuencias de los genes hsp20 y hsp70 de 42 cepas de diferentes orígenes geográficos, que representan 14 especies de Leishmania, se concatenaron y analizaron como mismo se realizó el análisis filogenético previo basado en el gen hsp70. De forma general se corroboraron los resultados obtenidos con el gen hsp70, con la excepción de que L. (L.) aethiopica y L. (L.) tropica se soportan como dos grupos distintos y pueden considerarse como dos especies separadas. De destacar, además, se corroboraron los resultados previos obtenidos donde hay una separación de L. infantum y L. peruviana como subgrupos dentro de los complejos L. (L.) donovani y L. (V.) braziliensis, respectivamente. 23
Como resultados adicionales podemos señalar que por primera vez el gen de la HSP20 fue utilizado en estudios filogenéticos del género Leishmania, constituyendo un marcador molecular útil para este tipo de estudios. 23
El análisis combinado de los genes hsp70+ 7 Spliced-Leader RNA (7SL-RNA)+ espaciadores internos de la transcripción (ITS) ARN ribosómico (ARNr) (rDNA ITS1)+ mini exón refuerza los resultados obtenidos con el gen hsp70 (datos no mostrados). Los mismos grupos monofiléticos fueron encontrados con el análisis concatenado con la con la excepción de que L. (L.) aethiopica y L. (L.) tropica, aun cuando se soportan en un grupo monofilético con alto valor de ¨bootstrap¨ se soportan como dos grupos distintos y pueden considerarse como dos especies separadas, así como se había descrito anteriormente al combinar los genes hsp70+hsp20.
Como se ha presentado el análisis filogenético del gen hsp70 agrupa a las especies del género Leishmania de mamíferos en ocho grupos monofiléticos. Los grupos corresponden a especies, o a los llamados complejos de especies. Cada uno de los ocho grupos obtenidos es discutido en detalle a continuación, junto con una vista alternativa sobre definiciones de especies basadas en el gen hsp70 y el análisis concatenado.
Complejo L. (L.) donovani: Este grupo comprende todas las especies causantes de leishmaniasis visceral (L. donovani, L. archibaldi, L. infantum, y L. chagasi). En la Figura 2 queda claro que L. infantum y L. chagasi no pueden distinguirse una de la otra y comprenden un subgrupo dentro del grupo L. donovani. Estos resultados están de acuerdo con el análisis de microsatélites, RFLP, RAPD y secuenciación de ADN y esto es consistente con la reciente introducción de L. infantum en el Muevo Mundo 2,24-26. Por su parte L. archibaldi se ha cuestionado como un taxón independiente ya que diferentes marcadores moleculares no lo distinguen de L. donovani27,26,28-32. Además, sobre la base de la hsp70 no pueden ser diferenciadas. Sobre la base de estos datos y los análisis combinados realizados recomendamos reconocer a L. donovani como una única especie en el complejo L. donovani con L. donovani infantum como una subespecie.
Complejo L. (L.) tropica: Basado en el gen hsp70, L. tropica no se puede distinguir de L. aethiopica, ambas especies forman un mismo grupo con alta fidelidad estadística. Estos resultados han sido obtenidos mediante el análisis filogenético de otros marcadores moleculares 27,33,34,35,36,38. En función de estos resultados y como ninguno de los grupos es monofilético, no se le puede dar el estatus de subespecie. De ahí que solo se reconozca una sola especie L. tropica basado en estos resultados. Sin embargo, al realizar los análisis concatenados estas dos especies aparecen como entidades separadas con alto valor de “bootstrap”, aun cuando queden agrupadas por lo que deben ser consideradas como dos especies L. tropica y L. aethiopica.
L. (L.) major: Lainson y Shaw 1 incluyeron a L. major dentro del complejo L. tropica. Sin embargo, basado en el gen hsp70 claramente se identifica como un grupo monofilético separado de L. tropica como también se observó mediante el análisis por MLEE 2, secuencia repetitiva ADN 38, cytB 35, 7SL RNA 37, y la región ITS 33-34. Nuestros resultados soportan el estatus de L. major como una especie independiente como propuso la OMS 37 y Bañuls et al. 3.
Complejo L. (L.) mexicana: Tres especies quedan agrupadas en este complejo L. amazonensis, L. mexicana y L. garnhami. Este grupo está muy bien definido como un grupo diferente a los grupos correspondientes a las especies del subgénero Leishmania del Viejo Mundo (figuras 1 y 2). Resultados similares fueron obtenidos por el análisis de diferentes genes 33,35-37,39. En nuestro análisis ninguna de las 3 especies puede distinguirse como clados independientes. De ahí que no podamos definir como subespecies basado en el gen hsp70, y reconozcamos a L. mexicana como la única especie reconocida. Aunque el análisis combinado parece diferenciar L. amazonesis-L. garnahami de L. mexicana en dos grupos bien definidos, sin embargo, se necesitan un mayor número de cepas representantes de las 3 especies para llegar a conclusiones al respecto.
L. (V.) naiffi: Esta es una de los cuatro grupos reconocidos dentro del subgénero Viannia, y que está de acuerdo con observaciones previamente obtenidas por diferentes autores 3,40,41. Pocos estudios han analizado el estatus filogenético de como especie y en todos los casos se han utilizado muy pocos aislamientos, con resultados similares 42-45. Como en nuestro estudio solo se pudo utilizar un aislado de esta especie, no podemos investigar su estado monofilético. Aun cuando con los análisis concatenados donde se usaron entre 2 y 3 aislamientos este parece ser un clado independiente.
Complejo L. (V.) braziliensis: Este grupo de forma por dos especies L. braziliensis y L. peruviana. Algunos autores sugieren que L. peruviana es simplemente una variante de L. braziliensis y no una especie validada 46. Estos resultados son soportados por nuestro estudio. Como se observa en la Figura 2, L. peruviana aparece como un subgrupo dentro de la población de L. braziliensis, resultado también obtenido mediante el análisis concatenado. De ahí que se sugiera basado en el gen hsp70 el estatus de especie para L. braziliensis para todos los aislamientos de este complejo con L. braziliensis peruviana como una subespecie.
Complejo L. (V.) guyanensis: Como se encontró previamente con el análisis de isoenzimas 2,24,40, patrones de restricción del gen ITS ADNr 42, y el análisis filogenético de ITS ADNr y cytB 33,35, L. guyanensis y L. panamensis forman un grupo monofilético (figura 1). Dentro de este complejo, ambas especies forman sub-clúster monofiléticos, confirmando los resultados de una amplia gama de isoenzimas y marcadores de RAPD 47, así como el análisis filogenético basado en ITS ADNr 34,43,45) que muestran una separación clara. En conclusión, los datos basados en el gen hsp70 data sugieren identificar a L. guyanensis, con L. guyanensis panamensis como una subespecie.
L. (V.) lainsoni: Esta especie presenta las más distintivas características biológicas (morfología, crecimiento en medio de cultivo exentico), bioquímicas (patrones de electroforesis enzimáticas), y de biología molecular dentro del subgénero L. (Viannia). Nuestro análisis basado en el gen hsp70 mostro a L. lainsoni como la especie más divergente dentro del subgénero L. (Viannia) (Figura 1). Nuestros datos identifican a L. lainsoni como una especie separada dentro del subgénero L. (Viannia) como se describió anteriormente 3,24,42,48) y se corrobora con los resultados de los análisis concatenados.
De forma general proponemos entonces simplificar la clasificación actual taxonómica de Leishmania basado en los grupos monofiléticos encontrados en base al gen hsp70 (tabla 1).
Tabla 1 Nomenclatura simplificada en el análisis del gen hsp70
| Subgénero (localizacion intestino vector) | Complejo (Grupo de especies/estatus especie cuestionados) | Especies (Vector, distribución geográfica, tropismo, propiedades antigénicas y manifestaciones clínicas) | |
|---|---|---|---|
|
|
|||
Por primera vez el gen de la HSP70 citoplasmática fue utilizado en estudios filogenéticos del género Leishmania y con los resultados obtenidos se demostró su utilidad para realizar estos análisis. Este estudio contribuyó significativamente a promover la discusión sobre un marco de consenso taxonómico para Leishmania.49
Proponemos abandonar el concepto de complejos de especies, ya que ninguno de ellos parece estar compuesto de entidades monofiléticas separadas. Proponemos además simplificar la clasificación taxonómica de Leishmania, basado en los resultados filogenéticos basados en función de la hsp70 en función de lograr que sea fácilmente aplicable desde un punto de vista clínico y epidemiológico para el beneficio de pacientes y personas en riesgo. 50,51
Actualmente la clasificación del Género Leishmania se basa en los resultados obtenidos basados en el gen de la hsp70.4,50,51
Origen del género Leishmania
Hoy existen tres hipótesis para los orígenes del género Leishmania: Paleártica, Neotropical y Múltiples orígenes. En la hipótesis Paleártica, se asume el origen de Leishmania de lagartos cretáceos con migraciones recientes al Neártico, lo que sugiere que Sauroleishmania forma un clado hermano para todas las demás especies. En contraste, la segunda hipótesis, sugiere el origen de la Leishmania en el Neotrópico, respaldado por filogenias basadas en secuencias. Aquí los datos sugieren que las especies del nuevo mundo surgieron hace 46-34 millones de años y son los antepasados de las especies del viejo Mundo, haciendo de Sauroleishmania una forma derivada de mamíferos y no ancestral. Por último y no menos importante, la hipótesis del origen múltiple sugiere una división de Leishmania en dos linajes de Euleishmania (incluidos los subgéneros de Leishmania, Viannia y Sauroleishmania) y Paraleishmania en el supercontinente Gondwana. Posteriormente la ruptura de Gondwana separó a los antepasados de todos los subgéneros de Leishmania.52
El análisis realizado basado en el gen hsp70 y en la combinación de los genes estudiados proporciona evidencia convincente en apoyo de la hipótesis de que el género Leishmania puede haberse originado en América del Sur (Nuevo Mundo), apoyando la hipótesis Neotropical, dentro de las tres hipótesis que existen para los orígenes del género, en concordancia con otros estudios 1,10,20,26,37,38. En cada uno de los árboles filogenéticos presentados (figura 1, 2) las ramas más ancestrales corresponden a las especies del subgénero L. (Viannia) que incluyen aquellas especies que se encuentran distribuidas en América del Sur. De forma general se muestra que las especies del Nuevo Mundo se ramifican cerca de la base de los árboles y las especies del Viejo Mundo en la corona del subgénero L. (Leishmania).
Nuestro análisis proporciona evidencias que soportan la hipótesis de que el género Leishmania fue originado en América del Sur (Nuevo Mundo), contribuyendo a la discusión general sobre el origen de este género.
Análisis filogenético del género Trypanosoma
La figura 3 muestra el árbol construido a partir de las secuencias nucleotídicas utilizando el método de Unión al Vecino “Neighbor-Joining”. Nueve grupos son reconocidos: T. carassi, Trypanozoon, T. congolense, T. vivax, T. theileri, T. grayi, T. lewisi, T. rangeli y T. cruzi. El clado Trypanozoon incluye T. brucei gambiense, T. b. rhodesiense, T. b. brucei, T. evansi y T. equiperdum. El subgénero Megatrypanum no es compatible como la sección Stercoraria. El subgénero Herpetosoma forma un grupo parafilético, con el subgénero Schizotrypanum ramificándose entre T. rangeli y T. lewisi. El grupo T. cruzi incluye T. cruzi y T. cruzi marinkellei. Este último como un subgrupo dentro del clado T. cruzi.14
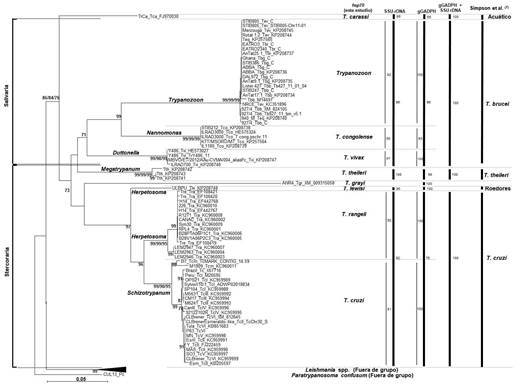
Fig. 3 Árbol filogenético basado en las secuencias del gen hsp70 de 11 especies de Trypanosoma según el método de Unión al Vecino (Neighbor-Joining). La distancia fue estimada por el método de Kimura 2-parameter. El análisis de “bootstrap” 2000 réplicas. Especies del viejo Mundo se representan con círculos en la rama, mientras que especies del nuevo Mundo con un cuadrado. El árbol está enrutado con las especies de Leishmania y Paratrypanosoma confusum. Los nombres de las ramas representan la clasificación clásica de subgéneros de Hoare 5. Las secciones Stercoraria y Salivaria se indican a la izquierda.
Estos grupos fueron también verificados en árboles filogenéticos construidos por los métodos de Máxima Parsimonia y Máxima Verosimilitud, así como los árboles obtenidos basados en las secuencias aminoacídicas deducidas 14.
Utilizando las mismas secuencias nucleotídicas se construyó una red filogenética utilizando el método de “Neighbor-Net”, modelo “p-distance” no corregido. Como se puede observar en la figura 4A, los mismos grupos que se identificaron por el análisis convencional se muestran en esta red. En el clado T. rangeli se pueden distinguir dos grupos (figura 4B). Las especies o subespecies dentro del subgénero Trypanozoon no se pueden distinguir (figura 4C). Se observan cuatro grupos genéticos en T. cruzi, correspondiente a las Unidades Discretas de tipificación (DTU): TcI, TcIII, TcIV y los DTUs TcII, TcV y TcVI que no forman entidades separadas (figura 4D). 14
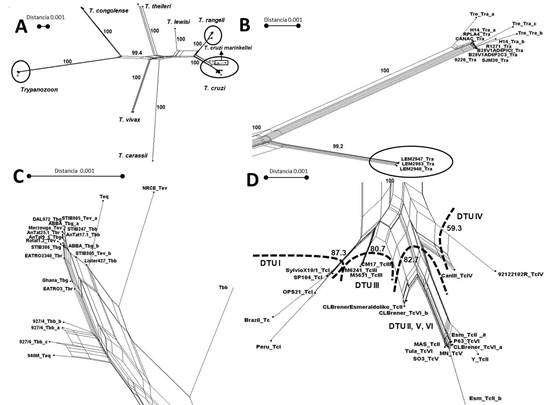
Fig. 4 Red filogenética basada en las secuencias del gen hsp70 de distintas especies de Trypanosoma, según el método “NeighborNet algorithm” cada uno de los cuatro paneles (A)- (D) se dibuja a la escala indicada. (A). Red completa con representaciones de los tres grupos mostrados con más detalles en los paneles restantes. (B.) Clado T. rangeli (C). Clado Trypanozoon (D). Clado T. cruzi. Dentro del clado se diferencias las DTU.
El análisis combinado de subunidad menor ARNr (SSU-ADNr)+ gliceraldehido 3´fosfato deshidrogenasa (gGADPH) +hsp70 resuelven aún más las relaciones filogénicas dentro del género Trypanosoma (datos no mostrados). El análisis fue consistente con el análisis de del gen hsp70, con pocas excepciones. Primero, T. theileri se agrupó consistentemente con la sección Stercoraria con una alta fidelidad en todos los árboles. Segundo, T. vivax claramente se agrupa con el clúster Trypanozoon-T. congolense.14
Como se ha presentado el análisis filogenético basado en el gen hsp70 agrupa a las especies del género Trypanosoma en nueve grupos monofiléticos. Los grupos corresponden a especies o subespecies. Cada uno de los grupos obtenidos es discutido en detalle a continuación.
Clado Trypanozoon: El subgenera Trypanozoon forma un grupo monofilético constituido por las subspecies T. brucei brucei, T. b. rhodesiense, T. b. gambiense, y las especies T. evansi y T. equiperdum) que no se soportan como grupos separados. Ni por el análisis por el gen hsp70, ni por el análisis concatenado con otros marcadores genéticos se obtienen diferencias en este grupo. La monofilia de Trypanozoon se ha observado además con diferentes marcadores moleculares. 9,53-55T. evansi y T. equiperdum por lo tanto, no son clados monofiléticos y no califican para el estado de especie.
Diferentes autores proponen incluir dentro del clado T. brucei a las especies T. congolense, T. vivax así como otras especies trasmitidas por la mosca tsetse (Trypanosoma simiae y Trypanosoma godfreyi), basado en los resultados obtenidos por el análisis filogenético de los genes SSU-rRNA y gGADPH.9,10,56-59 Aun cuando en nuestro análisis T. brucei, T. congolense, y T. vivax se agrupan juntos en un mismo clado. Sin embargo, en todos los análisis, T. congolense y T. vivax forman entidades distintivas con altos valores de ¨bootstrap¨ y por ende consideramos que deben ser consideradas especies separadas en los clados T. congolense y T. vivax. (figura 3).
Clado T. theileri: En nuestro análisis basado en el gen hsp70 solo, T. theileri no se une convincentemente con la sección Stercoraria sino con la Salivaria. Esta posición dentro del género Trypanosoma ha sido muy debatida y hasta el momento no se han podido establecer claramente las relaciones. Algunos estudios demuestran que T. theileri está más cerca de T. cruzi que de T. brucei basado en el estudio filogenético de los genes SSU-rRNA y gGAPDH 54,59-61. Sin embargo, el análisis combinado realizado en este estudio basado en los genes SSU-rDNA+gGADPH+hsp70 agrupa a T. theileri con la sección Stercoraria. Sin embargo, otros estudios han mostrado resultados contradictorios basados en los mismos genes 56,58. Nuestros resultados soportan a T. theileri como un clado independiente como propusieron Simpson et al.; 7 Rodrigues et al. 61) y Hamilton et al.60
Clado T. grayi: Basado en el análisis filogenético del gen hsp70 solo, T. grayi aparece como un taxón independiente, no relacionado claramente con ningún grupo en nuestro análisis. Sin embargo, en el análisis concatenado SSU-rDNA + gGADPH + hsp70 las secuencias claramente se agrupan más relacionadas con T. cruzi que con el clado T. brucei. Kelly et al. 62 concluyeron que T. grayi está más relacionado con T. cruzi que con T. brucei, pero el análisis filogenético con SSU-rRNA y gGAPDH lo ubica en un clado separado de T. cruzi y T. brucei, a menudo con otros tripanosomas de reptiles o aves. 9,59,62
Clado T. lewisi: Dentro del subgénero Herpetosoma se encuentran las especies T. lewisi y T. rangeli. Sin embargo, de acuerdo a los resultados obtenidos basados en el gen hsp70, este subgénero es claramente parafilético. Añez 63) propuso eliminar a T. rangeli del subgénero Herpetosoma, creando un Nuevo subgénero Tejeraia. Otros autores sugieren su reclasificación en el subgénero Schizotrypanum, o descontinuar el uso de subgéneros basado en los resultados filogenéticas realizados con los genes SSU-rRNA, gGAPDH, proteasa tipo catepsina L, cyt b, y SL RNA. 10,51,64-66
Clado T. rangeli: El análisis basado en el gen hsp70 identifico a T. rangeli como un clado hermano a T. cruzi y T. cruzi marinkellei, al igual que al utilizar otros marcadores moleculares. 9,10,64-66
Clado T. cruzi: El análisis del gen hsp70 y el análisis concatenado basado en los genes SSU rDNA+gGADPH+hsp70 soportan que el clado T. cruzi clade como describió Simpson et al. 59 (figura 3). T. cruzi es una de las especies del subgénero Schizotrypanum que constituye un grupo heterogéneo monofilético dentro del género Trypanosoma.5,67 Se acepta actualmente que al menos existen seis Unidades Discretas de tipificación (DTUs) 68: TcI-TcVI.67,69 Cuatro grupos fueron soportados mediante el gen hsp70 en la red filogenética (Figura 4D) correspondiente a TcI, TcIII, TcIV y TcII + V + VI. Como TcV y TcVI son resultado del evento de hibridación de TcII y TcIII, siendo este el más cercano proceso de hibridación a los que se ha sometido esta especie, 70-73 no nos sorprende las relaciones entre estos linajes especialmente con TcII. 73-74 Basado en el gen hsp70 no es posible discriminar los DTUs TcII, TcV, y TcVI. Dos genotipos relacionados con T. cruzi están relacionados con muercielagos: Tc-bat y T. cruzi marinkellei.75-76 La genealogía de los genes hsp70 inferidos en este estudio confirma que los aislados de T. cruzi marinkellei son un clado hermano de los aislados de T. cruzi humanos, ambos formando grupos monofiléticos (figura 3). Estos resultados están en concordancia con los previamente reportados con diferentes los genes, 56,65,75 y por el análisis de proteómica comparativa. 77
Por primera vez el gen de la HSP70 citoplasmática fue utilizado en estudios filogenéticos del género Trypanosoma y con los resultados obtenidos se demostró su utilidad para realizar estos análisis. La filogenia de Trypanosoma derivada del análisis del gen hsp70 se suma a la evidencia existente hacia la clasificación de este género en clados filogenéticas y descontinuar la clasificación clásica de sección y subgénero.
Conclusiones
Se depositaron en el Banco de Genes la secuencia parcial del gen hsp70 citoplasmático de 43 cepas de 11 especies de Leishmania y 41 de 9 especies de Trypanosoma, lo que permitirá realizar estudios futuros y que contribuye al conocimiento de la comunidad científica internacional. El análisis del polimorfismo genético basado en la secuencia del gen hsp70 demostró que este marcador molecular es útil para ser utilizado en el diseño de métodos moleculares para la detección, identificación y diferenciación de especies y linajes de Leishmania y Trypanosoma.
Por primera vez el gen de la HSP70 citoplasmática fue utilizado en estudios filogenéticos de los géneros Leishmania y Trypanosoma, este gen se encuentra bajo una selección purificadora que le permite su conservación y preservar la funcionalidad de la proteína y con los resultados obtenidos se demostró su utilidad para realizar estos análisis. Mediante la secuenciación de locus-simple, basado en el gen hsp70, se puede realizar la identificación de especies de Leishmania y Trypanosoma sin la necesidad del aislamiento del parásito. Con este estudio, se contribuyó activamente a la discusión sobre un marco general de consenso taxonómico para los géneros Leishmania y Trypanosoma.
El concepto de complejos de especies para agrupar especies de Leishmania basado en características biológicas y bioquímicas no está respaldado por filogenias moleculares y proponemos descontinuar la clasificación clásica de sección y subgénero de las especies de Trypanosoma y proponer la clasificación en clados, ya que las secciones y subgéneros son claramente polifiléticos. Nuestro análisis proporciona evidencias que soportan la hipótesis de que el género Leishmania fue originado en América del Sur (Nuevo Mundo), contribuyendo a la discusión general sobre el origen de este género.
Los resultados basados en el análisis filogenético del gen que codifica la hsp70 fueron tomados en cuenta y sirvieron de base para la actual clasificación taxonómica de Leishmania, que busca el mayor grado posible de relevancia práctica, bajo los principios de reducción, simplificación y delineación, en función de lograr que sea fácilmente aplicable desde un punto de vista clínico y epidemiológico para el beneficio de pacientes y personas en riesgo.

