Descriptores DeCs: DELECCIÓN CROMOSÓMICA; ANOMALÍAS CROMOSÓMICAS/genética; CROMOSOMAS HUMANOS PAR 18/genética; TRANSLOCACIÓN (GENÉTICA)/genética; CARIOTIPIFICACIÓN; FENOTIPO; LINAJE.
La monosomía parcial 18q-, también conocida como síndrome de Grouchy, fue descrita por su autor en 1964.1 Su defecto básico es un raro trastorno cromosómico que consiste en la delección de parte del brazo largo del cromosoma 18, que afecta la banda 18q 21;2 no obstante, un paciente con el síndrome completo fue descrito con una delección q 22-ter.3
A la primera descripción, le siguieron otras: Lejeune en 1966 y una revisión de Schinzel en 1975.4 Hasta el momento se han publicado alrededor de 80 casos y se ha planteado que en el 75 % no hay afectación de los padres, por tratarse de una cromosomopatía de "novo" que afecta uno y otro sexos y puede, ocasionalmente, proceder de un padre con mosaicismo.4 Se ha observado translocación en el 15 % de los pacientes.2
En cuanto a la gravedad del cuadro clínico parece no afectar la sobrevivencia, pero es grave la oligofrenia,5 y el paciente de mayor edad que se ha descrito tenía 20 años.2 No se ha informado que exista asociación con la edad avanzada de los padres.2
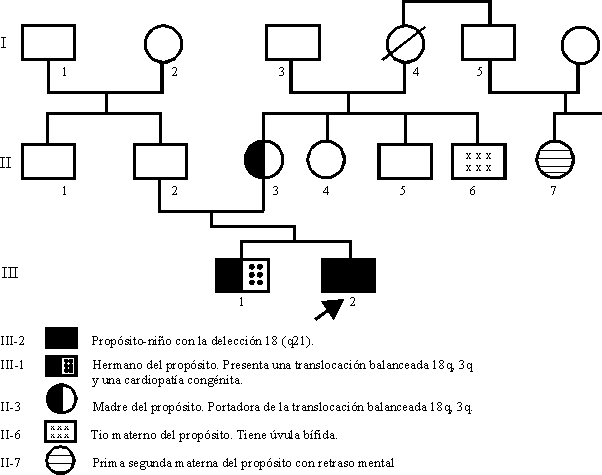
El hallazgo, en un niño de 4 años con retraso mental y anomalías congénitas menores de un cariotipo 46 XY18q-, nos motivó a profundizar en la correlación fenotipo-cariotipo de la monosomía 18q- y en la importancia del estudio citogenético y del examen físico minucioso a los padres de niños con alteraciones cromosómicas estructurales.
El embarazo del propósito fue normal; la madre ingirió alcohol ocasionalmente y mucho café. A las 39 semanas ocurrió rotura de membrana; se indujo el parto con oxitocina, el cual fue eutócico, y se obtuvo un recién nacido de 2 500 g, sin alteraciones aparentes. Según referencias de la madre, el desarrollo psicomotor durante el primer año fue bueno, pero comenzó a caminar al año y medio y no habló.
Al realizar examen físico en el momento de la remisión se comprueba que el niño no habla, y se comunica mediante gestos; presenta circunferencia cefálica de 49 cm (25 P), talla 94 cm (10 P), pelo rubio casi blanco y seco, branquicefalia, abombamiento frontal, ojos hundidos, epicanto, hipoplasia del macizo facial medio, puente nasal deprimido y ancho, nariz pequeña, labio superior fino, prognatismo y orejas grandes, cuello corto, piel seca y gruesa, extremidades con dedos largos, sindactilia II-III en dedos de ambos pies, limitada a la falange proximal. Genitales externos: prepucio que no cubre glande, bolsas escrotales pequeñas.
Dermatoglifo: Trirradio axial en posición distal (t") en mano izquierda, y un pliegue transverso distal en mano derecha.
Dosificación de IgA: no útil. ]]>
Estudio audiométrico: normal.Examen físico del hermano del propósito: Niño de 7 años de edad, talla 177 cm (25 P), ligero prognatismo, orejas discretamente grandes, se refiere que no hay dificultades en el aprendizaje. Cardiopatía congénita.
Al examen físico, la madre del propósito presenta hipertelorismo, ojos profundos, discreto prognatismo y orejas grandes.
El examen físico del padre del propósito no arroja datos de interés.
Al realizar el estudio citogenético a la familia por bandas G(GTG),6 se obtuvieron los siguientes resultados:
Además de estos criterios, la literatura médica recoge un grupo importante de anomalías, de modo que el fenotipo es bastante constante; por ello, quien ha visto una vez el síndrome, puede reconocerlo clínicamente; no obstante, el cariotipo es indispensable para determinar el diagnóstico de certeza, y el diagnóstico diferencial habría que establecerlo con los síndromes de Apert, Crouzon y Fraccaro, y con el retraso mental moderado o importante de causa desconocida.5 Recientemente se ha descrito la delección intersticial del brazo largo en 5 pacientes, lo que pudiera constituir una entidad clínica distintiva.8,9
En el anexo se muestran de forma resumida todos los hallazgos informados sobre este síndrome en la literatura médica revisada, y se especifica cuáles estaban presentes en nuestro paciente.
Los hallazgos clínicos de anomalías menores que constituyen un patrón dismórfico facial en la madre y hermano del propósito, portadores aparentemente balanceados de la translocación, es una prueba más de que un elevado porcentaje de los individuos con translocaciones equilibradas tienen expresión clínica o subclínica y son mentalmene subnormales.10 Ello justifica el criterio de que, al menos los familiares de primer grado, deben ser rutinariamente examinados y que el cariotipo en sangre periférica estará siempre indicado en los padres y hermanos de niños con alteraciones cromosómicas estructurales, como es la delección terminal 18q.
No tenemos suficientes evidencias para afirmar que la cardiopatía congénita presente en el primogénito forme parte de las consecuencias de la translocación 3q, 18q, y puede ser una asociación fortuita, aunque se han informado defectos cardíacos ocasionales en el síndrome 18q-.11
Respecto a los hallazgos reportados en relación con el desarrollo de enfermedades autoinmunes, anemia perniciosa, hipotiroidismo, deficiencia de IgA y tumores malignos, como astrocitoma cerebral y tumor de mediastino en pacientes con síndrome 18q-, los criterios en la literatura médica todavía no son concluyentes y la edad de aparición es a veces muy tardía; pero se aportan paulatinamente elementos sobre la interrelación de la genética y la oncogénesis.12 Es importante señalar el criterio de varios autores de que entre los síndromes clínicos 18q- hay algunos que en realidad son 18 r, y por tanto presentan también delección 18p-. Esta última ha sido observada en asociación con la dismorfogénesis tiroidea, por lo que podría constituir en realidad una entidad independiente.13 La dosificación de IgA en nuestro paciente no fue útil.
En conclusión, creemos que nuestro paciente presenta un cuadro clínico que se puede considerar típico dentro de las monosomías 18q-. El hallazgo de una translocación heredada en nuestro caso, demuestra la importancia de estudiar a los padres de niños con retraso mental y anomalías menores, tanto desde el punto de vista citogenético como clínico.
| Hallazgo clínico | | |
| 1. Peso al nacimiento menor de 3 000 g | ]]> 2,4,7,11 | |
| 2. Estatura pequeña | | |
| 3. Deficiencia mental marcada | | |
| 4. Hipotonía | | |
| 5. Pobre coordinación | | |
| 6. Nistagmo | ]]> 5,4,7,11 | |
| 7. Sordera conductiva o hipoacusia | | |
| 8. Microcefalia moderada | | |
| 9. Hipoplasia mesofacial | | |
| 10. Ojos hundidos | | |
| 11. Boca de carpa | ]]> 5,2,4,7,11 | |
| 12. Dedos largos, en palillo de tambor | | |
| 13. Implantación proximal pulgar | | |
| 14. Anomalías dermatoglíficas (exceso de espiras, pliegue simiano) | | |
| 15. Talipes equinovaro (talus vertical) | | |
| 16. Anomalías genitales (criptorquidia, micropene, escrotos pequeños) | ]]> 5,2,4,11 | |
| 17. Hoyuelos sobre articulaciones y mejillas (nódulo subcutáneo) | | |
| 18. Hipertelorismo | | |
| 19. Epicanto | | |
| 20. Defectos de retina, córnea, disco óptico. | | |
| 21. Convulsiones al nacimiento | ]]> 2,4 | |
| 22. Frente abombada | | |
| 23. Prognatismo | | |
| 24. Cuello corto | | |
| 25. Pezones separados | | |
| 26. Cardiopatías congénitas | ]]> 2,4,11 | |
| 27. Hernia umbilical | | |
| 28. Cierre tardío de las fontanelas | | |
| 29. Pelo anormal (escaso, seco, alopecia) | | |
| 30. Glaucoma, estrabismo, catarata | | |
| 31. Nariz corta, puente nasal ancho | ]]> 2,4 | |
| 32. Orejas anormales (antitrago y antihélix prominente) | | |
| Hallazgo clínico Autores Paciente | ||
| 33. Labio leporino ± paladar hendido | | |
| 34. Disminución de la IgA | | |
| 35. Costillas supernumerarias | | |
| 36. Malformaciones renales | ]]> 4,5 | |
| 37. Orejas de baja implantación | | |
| 38. Eccema | | |
| 39. Conducta autista | | |
| 40. Posición anormal de los dedos de los pies. | | |
| 41. Voz ronca, sin tono | ]]> 11 | |
Subject headings: CHROMOSOME DELECTION; CHROMOSOME ABNORMALITIES/genetics; CHROMOSOMES HUMAN, PAIR 18/genetics; TRANSLOCATION (GENETICS)/genetics; KARYOTYPING; PHENOTYPE; PEDIGREE.
Dra. Manuela Herrera Martínez. Calle 8va, edificio 2, apartamento 7, entre 9na y Circunvalación, barrio Escambray, municipio Santa Clara, Villa Clara, Cuba.
1 Especialista de II Grado en Genética Clínica. Asistente. Facultad de Medicina. Instituto Superior de Ciencias Médicas de Villa Clara (ISCM-VC).
2 Especialista de I Grado en Genética Clínica. Asistente. Facultad de Medicina. ISCM-VC. ]]>