Variantes de la heterocromatina y la eucromatina en el diagnóstico prenatal citogenético
Variants of heterochromatin and euchromatin in cytogenetic prenatal diagnosis
MSc. Michel Soriano-Torres, MSc. Enny Morales Rodríguez, Dra. Iris Rojas Betancourt , Dr. C. Luis Alberto Méndez Rosado ]]>
Centro Nacional de Genética Médica. La Habana, Cuba.
RESUMEN
Algunos cambios en la morfología de los cromosomas, detectados durante el análisis citogenético, no están asociados con defectos clínicos, representan un dilema para el asesor genético principalmente durante la realización de un estudio prenatal; por esta razón es que una apropiada discriminación entre una variante inocua y una verdadera anomalía resulta crucial para llevar a cabo un asesoramiento genético preciso. Los polimorfismos de la heterocromatina son identificados usualmente por técnicas de bandeo específicas y consideradas como variaciones mendelianas sin una significación clínica. De igual modo, en la literatura se expone la presencia de variantes en regiones eucromáticas que después de un análisis detallado resultan ser de naturaleza benigna. Debido a la importancia de este tema en la actualidad se hace necesario proponer un protocolo a seguir en los laboratorios cada vez que una variante cromosómica sea detectada en el diagnóstico prenatal. El objetivo de este trabajo es presentar una revisión de la literatura acerca de los pasos que se siguen ante la aparición de una variante cromosómica y las sugerencias que se brindan para un manejo más adecuado.
]]>
Palabras clave: diagnóstico prenatal citogenético, variantes heteromórficas, variantes eucromáticas, heteromorfismo.
ABSTRACT
Some changes in chromosome morphology, which are detected in cytogenetic diagnostics, are not associated with clinical defects presenting a dilemma for the genetic counsellor, especially during prenatal diagnosis; this is the reason why a proper discrimination between innocuous variants and true anomalies is crucial to allow precise counselling. Polymorphisms of heterochromatin are identified usually by specific banding techniques and considered as Mendelian variations without a clinical significance. Likewise, it has been exposed in the literature the presence of variants in euchromatic regions that after a detailed analysis turns out to be of benign nature. Due to the current importance of this issue it is necessary to propose a protocol to follow in our laboratories every time a chromosome variant is detected while performing a prenatal analysis and supported by experienced specialist in our field. The goal of this work is to present a review of the literature about how a finding of a chromosome variant is handled and the suggestions given for a more proper management.
Keywords: cytogenetic prenatal diagnosis, heteromorphic variants, euchromatic variants, heteromorphism.
]]>
INTRODUCCIÓN
Cuando se lleva a cabo un estudio citogenético, en ocasiones, pueden detectarse algunas variaciones apreciables en la morfología de los cromosomas que no tienen un efecto adverso en el fenotipo del paciente. Estas variaciones son conocidas como "heteromorfismos" y se encuentran presentes en regiones cromosómicas microscópicamente visibles donde el tamaño, la morfología y las propiedades en la coloración pueden diferir entre cromosomas homólogos.1 Algunos de los heteromorfismos que suelen aparecer con más frecuencia se hallan en las regiones heterocromáticas de los cromosomas 1, 9, 16 y Y, al igual que en los satélites y los tallos de los cromosomas acrocéntricos.2
Para caracterizar este fenómeno suelen emplearse los términos: heteromorfismo, variante y polimorfismo. Sin embargo, el término polimorfismo se aplica mejor en el contexto de los genes y las moléculas, mientras que los términos variante cromosómica y heteromorfismo son más apropiados para describir las variaciones estructurales apreciables en los cromosomas mediante los métodos convencionales de la Citogenética.3
La edición del año 2005 del International System for Human Cytogenetic Nomenclature describe la nomenclatura a emplear para reportar las variaciones en los segmentos de heterocromatina, tallos satelitales y satélites y de este modo diferenciarlas de alteraciones estructurales de otra naturaleza (Shaffer y Tommerup, 2005). En la práctica diaria es común nombrar como variante cualquier deleción o duplicación una vez que son identificados otros miembros de la familia que portan el mismo desbalance y no presentan un fenotipo afectado.3
Resulta crucial para poder ofrecer un correcto asesoramiento genético lograr discriminar entre variantes cromosómicas y verdaderas anomalías. Lo anterior tiene especial significado durante la realización del diagnóstico prenatal citogenético (DPC) ya que la aparición de una variante estructural presenta un dilema para el especialista que realiza el diagnóstico.4 ]]>
Debido a la importancia que tiene esta temática en la actualidad a nivel internacional, se considera necesario desarrollar una propuesta de pasos a seguir en los diferentes laboratorios del país cuando se detecte una variante cromosómica durante el DPC, para optimizar la realización de estudios complementarios a los padres. Esta temática ha sido abordada con anterioridad en los talleres nacionales de Citogenética que han tenido lugar en los años 2008 y 2012; sin embargo, debido a la brevedad del espacio disponible para el debate científico y la diversidad de opiniones, no se ha logrado un acuerdo que sea de la satisfacción de todos. Este trabajo tiene como objetivo compilar la información que aparece en la literatura internacional sobre cómo se aborda la aparición de una variante cromosómica y las sugerencias hechas por los investigadores acerca de su manejo, para que pueda emplearse como base a la implementación de una metodología común en la red de servicios de Genética Médica.
DESARROLLO
Variantes heterocromáticas
Durante el análisis citogenético de rutina son detectadas frecuentemente variaciones morfológicas en la heterocromatina constitutiva de los cromosomas no acrocéntricos, estas variaciones en el tamaño y posición de la heterocromatina ocurre muy a menudo en la heterocromatina para/pericéntrica de los cromosomas 1, 9, 16 y en la heterocromatina distal del cromosoma Y.5,6 Las variantes pericentroméricas constituyen un dilema en el diagnóstico y en el asesoramiento, especialmente en los estudios prenatales, ya que es difícil realizar una distinción entre una alteración patogénica y una variante eucromática.7 ]]>
Las variaciones morfológicas del cromosoma 9 constituyen el segundo heteromorfismo más frecuente en los seres humanos después de aquellos que involucran los cromosomas acrocéntricos. Estas variaciones incluyen mayormente cambios en la extensión de la heterocromatina pericentromérica o la inversión de la región heterocromática pericentromérica, entre las bandas p11 y q13.8 Igualmente puede presentarse una banda eucromática extra insertada en la heterocromatina de la región 9q, esta fue reportada por primera vez en 1978 y luego caracterizada como una secuencia derivada a partir de la banda 9p12 mediante la técnica de fluorescencia in situ usando hibridación, más comúnmente conocida como FISH por sus siglas en inglés.7,9 Esta puede segregar en las familias sin consecuencias fenotípicas siendo recurrente en algunas poblaciones.4,10,11
La variación en la heterocromatina del cromosoma Y con tinción positiva por bandeo C estándar puede ir desde su virtual ausencia, haciéndolo comparable con la mitad del cromosoma 22, hasta una cantidad grande en cuyo caso puede alcanzar el mismo tamaño que el cromosoma 13. Realizar un estudio cromosómico paterno puede ser necesario para confirmar que un cromosoma muy pequeño es en verdad una variante, ya que si ocurrió una deleción y el punto de ruptura se encuentra proximal a la heterocromatina de la región Yq11-12 esta se considera patológica. En el caso de los cromosomas Y muy largos, resulta adecuado realizar un bandeo C para confirmar que el material adicional es heterocromatina ya que estudios citogenéticos realizados en más de 10 000 individuos han sugerido que los cromosomas Y largos están asociados con pérdida fetal. Una variante que suele presentarse comúnmente asociada al cromosoma Y es una inversión pericéntrica que le da una apariencia de cromosoma metacéntrico semejante a un cromosoma 20 de menor tamaño.12,13
En el caso de los cromosomas acrocéntricos, la presencia de material genético en los brazos cortos debe ser investigada realizando un estudio citogenético a ambos padres, ya que aunque a veces suele ser heterocromatina asociada a la región pericentromérica, también podría tratarse de material proveniente de un cromosoma no homólogo. La literatura internacional recoge el caso de un paciente síndrome de Beckwith-Wiedemann con una translocación desbalanceada entre los brazos cortos de los cromosomas 11 y 14, de este modo lo que fue interpretado en un inicio como una variante 14cenh+ (o 14p+) era en realidad una trisomía parcial de la región 11p causante de la afectación genética. Adicionalmente, en dos casos estudiados el material extra procedía de los cromosomas 6 y 19; lo anterior sugiere que un aumento sospechoso de material genético en los brazos cortos de cromosomas acrocéntricos debe ser corroborado mediante métodos moleculares apropiados, especialmente en estudios prenatales en los que el feto exhibe anomalías ultrasonográficas.14
Protocolos actuales de genética humana reconocen como variantes las siguientes: 1qh+, 9qh+, 16qh+, 18ph+, inv(1), inv(2), inv(9), inv(19), inv(Y), pstk+/pstk-, varYqh. De todas estas se menciona que son variantes cromosómicas, aparentemente sin relevancia clínica y bien conocidas.15
El College of American Pathologists y el American College of Medical Genetics aplicaron en el año 1997 una prueba de proeficiencia a 226 laboratorios de Citogenética. La prueba consistió en una encuesta sobre la apreciación por parte de sus miembros del carácter de variante o no de diversas variaciones estructurales que suelen aparecer con frecuencia en los cromosomas, y sobre si incluirían el hallazgo en el reporte oficial. Algunos heteromorfismos detectados mediante bandeo G estándar fueron considerados variantes normales por algunos participantes pero no por otros, el 61 % de los participantes expuso que ellos incluirían la información de los heteromorfismos hallados en el reporte clínico. Los brazos cortos prominentes, los satélites largos o dobles y el incremento en el largo de los tallos satelitales o tallos dobles en cromosomas acrocéntricos, fueron considerados como heteromorfismos por más del 90 % de los encuestados, pero solo una minoría (24 %~36 %) los incluirían en el reporte final. Respuestas similares se obtuvieron para las variaciones en la heterocromatina del brazo largo de los cromosomas 1, 9, 16 y Y; el 97 % de los participantes los consideraron heteromorfismos, pero únicamente el 24 % reportaría estos hallazgos. Sin embargo, la mayoría reportaría las inversiones pericéntricas aún considerándolas variantes normales, de igual modo reportarían otros heteromorfismos poco frecuentes.16 ]]>
Variantes eucromáticas
Se ha propuesto que todas las aberraciones cromosómicas desbalanceadas que no involucran regiones heterocromáticas y no implican un efecto fenotípico deben ser descritas como variantes eucromáticas y que este término debe quedar restringido a aquellas variaciones que son visibles a nivel citogenético.1,3 Varios reportes de la literatura muestran el hallazgo de variantes eucromáticas benignas en diferentes cromosomas, la mayoría afecta la región proximal de los brazos cortos,17 entre estas han sido descritas variaciones que involucran las regiones 8p23.1, 9p12, 9q13, 15q11.2 y 16p11.2.18-20
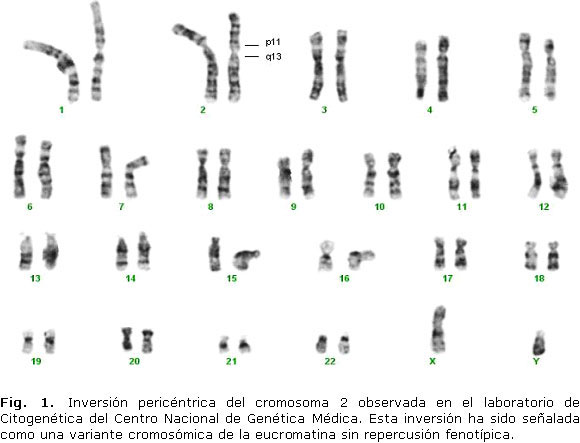
Algunas inversiones pericéntricas autosómicas son consideradas variantes polimórficas ya que se trasmiten de forma estable y no conllevan consecuencias fenotípicas.21 Algunas han sido estudiadas y están presenten en un alto porcentaje en determinadas poblaciones,22 un ejemplo de esto es la inv(2)(p11q13) que tiene una frecuencia de 0,1 % en los europeos del norte, es la inversión más común en humanos entre aquellas que no involucran a la heterocromatina centromérica (Fig. 1).23,24
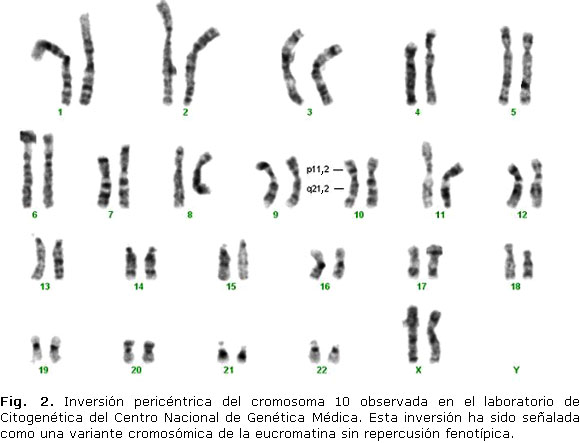
Otras variantes por inversión supuestamente no dañinas son las siguientes: inv(3)(p11q11), inv(3)(p11q12), inv(3)(p13q12), inv(5)(p13q13) y la inv(10)(p11.2q21.2), esta última puede apreciarse en la figura 2 y se considera una inversión pericéntrica común, ampliamente extendida geográficamente.5,24,25
Algunas regiones de eucromatina pueden estar delecionadas y no provocar un fenotipo adverso, este es el caso de la deleción de la región 13q21, la cual podría no conllevar ninguna repercusión fenotípica detectable.26-29 Es importante poder discriminar entre variantes eucromáticas y aberraciones patogénicas, ya que al menos un caso ha sido documentado donde una variante eucromática del 16p11.2 fue confundida con una duplicación patogénica, lo que conllevó a la terminación del embarazo.20
Se han reportado variantes en más de 50 regiones eucromáticas procedentes de casi todos los cromosomas autosómicos humanos. Actualmente se siguen reportando variaciones estructurales que no se correlacionan con ninguna expresión clínica aunque solo un grupo muy reducido de estos casos con variantes eucromáticas, ha sido caracterizado por medios moleculares.27,30,31 Distinguir duplicaciones patogénicas de variaciones eucromáticas es una tarea esencial en un laboratorio de Citogenética.32
CONCLUSIONES
Es importante tener en cuenta que existe un grupo de anomalías familiares donde el paciente posee alteraciones fenotípicas, mientras sus padres y cualquier otro portador en la familia son completamente sanos. En estos casos la anomalía puede influenciar el fenotipo del portador debido al fenómeno de impronta genómica, o es solo un hallazgo incidental y la verdadera causa de la anomalía en el fenotipo permanece sin ser descubierta.14 En la literatura se han reportado casos donde se sospechó en un inicio la presencia de una variante cromosómica, y el uso de técnicas avanzadas de Citogenética molecular, posteriormente reveló la presencia una aberración balanceada en uno de los progenitores. Es por esto que se debe ser cuidadoso con las aberraciones estructurales no consideradas relacionadas con manifestaciones clínicas, ya que una anomalía cromosómica más compleja responsable de defectos congénitos en el paciente puede estar presente, modificando ampliamente el asesoramiento genético. De hecho es aceptado, en la actualidad, que resulta esencial aplicar las nuevas tecnologías moleculares, como son la hibridación genómica comparativa mediante rearreglos, con el objetivo de caracterizar correctamente las alteraciones cromosómicas diagnosticadas años atrás con técnicas de menor resolución y de este modo identificar los verdaderos cambios benignos en el genoma, si los hay.33
]]>
Desafortunadamente, nuestra información limitada en cuanto a frecuencia y tipo de características clínicas asociadas con la detección prenatal de rearreglos aparentemente balanceados, no nos permite mejorar el asesoramiento genético prenatal actualizando el riesgo dado por Warburton en 1991.34 Es por esta causa que algunos autores recomiendan para la prueba diagnóstica de estas regiones con variantes eucromáticas, que dependiendo de la indicación clínica se realicen tanto la prueba citogenética como un análisis por microarreglos como pruebas complementarias.7 Sin embargo, a pesar de la disponibilidad de nuevas técnicas moleculares algunas técnicas convencionales como el bandeo C estándar y otros de la citogenética clásica no han dejado de aplicarse en el diagnóstico prenatal para caracterizar una posible variante cromosómica.17
Algunos autores han sugerido que las variantes deben ser registradas y también mencionadas a los pacientes en los reportes sin causar alarma, esto evitaría la repetición de este tipo de estudios en el futuro si se determinara que las variantes polimórficas juegan un papel en algunas condiciones clínicas específicas.12 Generalmente no existe una razón en particular para reportar una variante al médico que indica el estudio o al paciente, pero a veces es necesario realizar un estudio familiar cuando no queda claro si un hallazgo es una variante normal o una anomalía, o cuando un reporte previo ha provocado inseguridad en el paciente y este desea poseer más elementos sobre su estatus cromosómico.3
Es importante tener presente que los heteromorfismos citogenéticos son considerados como clínicamente insignificantes, pero determinar si un hallazgo cromosómico es verdaderamente un heteromorfismo o no, puede dificultarse. Por tanto, en ocasiones no siempre resulta posible realizar un manejo y asesoramiento genético apropiados especialmente cuando las variantes cromosómicas han sido halladas en pacientes evaluados con el objetivo de descartar una anomalía cromosómica.16
REFERENCIAS BIBLIOGRÁFICAS
1. Wyandt H, Tonk V. Normal population studies. In: Wyandt H, Tonk V, eds. Atlas of Human Chromosome Heteromorphisms. Dordrecht, The Netherlands: Kluwer Academic Publishers; 2004:33-46.
2. Sahin FI, Yilmaz Z, Yuregir OO, Bulakbasi T, Ozer O, Zeyneloglu HB. Chromosome heteromorphisms: an impact on infertility. J. Assist Reprod Genet. 2008;25(5):191-5.
3. Barber JC: Directly transmitted unbalanced chromosome abnormalities and euchromatic variants. J Med Genet 2005;42:609-29.
4. Eun Hae Cho, You Sun Kang, Eun Hee Lee. Extra G-Positive Band at Chromosome 9Q13 As a Recurrent Heteromorphism in a Korean Population. Fetal and Pediatric Pathology. 2011;30:257-9.
]]>
5. Gardner RJM, Sutherland GR. Variant Chromosomes and Abnormalities of No Phenotypic Consequence. In: Gardner RJM, Sutherland GR, editors. Chromosome Abnormalities and Genetic Counseling. 3th ed. Oxford University Press, USA; 2004. p. 233-48.
6. Hagymási K, Tulassay Z. The Human Genome Project, genetic viability and genetic epidemiology. Orv Hetil. 2005;146:2575-80.
7. Joseph-George AM, He Y, Marshall CR, Wong RCC, MacDonald JR, Fahey CA, et al. Euchromatic 9q13-q21 duplication variants are tandem segmental amplifications of sequence reciprocal to 9q13-q21 deletions. J Med Genet. 2011;48(5):317-22.
8. Verma RS. Heterochromatin: molecular and structural aspects. New York: Cambridge University Press; 1988. p. 276-299.
9. Wojiski SA, Harker Rhodes C, Brodhurst CA, Mohandas TK, Park JP. The G positive band of the rare euchromatic 9qh variant is derived from 9p12. Appl Cyto. 1997;23:125-8.
10. Fernández JL, Pereira S, Campos A, Gosálvez J, Goyanes V. An extra band within the human 9qh+ region that behaves like the surrounding constitutive heterochromatin. J Med Genet. 1994;31:632-4.
11. Starke H, Seidel J, Henn W, Reichardt S, Volleth M, Stumm M, et al. Homologous sequences at human chromosome 9 bands p12 and q13q21.1 are involved in different patterns of pericentric rearrangements. Eur J Hum Genet. 2002;10:790-800. ]]>
12. Madon P. Polymorphic variants on chromosomes probably play a significant role in infertility. Reproductive BioMedicine Online. 2005 [citado 7 Jul 2013];II(6):726-32. Disponible en: http://www.rbmonline.com/Article/1898
13. Salo P, Ignatius J, Simola KOJ, Tahvanainen E, Kaariinen H. Clinical features of nine males with molecularly defined deletions of the Y chromosome long arm. J Med Genet. 1995;32:711-5.
14. Kowalczyk M, Srebniak M, Tomaszewska A. Chromosome abnormalities without phenotypic consequences. J Appl Genet. 2007;48(2):157-66.
15. Lugon AHAD, Morton CCSV, Bieber FRYE, Fletcher JAZZ, Goersch ABIS, Kantarjian Spmi, etta aldi. Reporting of Diagnostic Cytogenetic Results. Current Protocols Human Genetics. Estados Unidos: John Wiley & Sons, Inc; 2001.
16. Brothman AR, Schneider NR, Saikevych I, Cooley LD, Butler MG, Patil S, et al. Cytogenetic heteromorphisms: survey results and reporting practices of giemsa-band regions that we have pondered for years. Arch Pathol Lab Med. 2006;(7):947-9.
17. Lecce R, Murdolo M, Gelli G, Stindl K, Coppola L, Romano A, et al. The euchromatic 9p+ polymorphism is a locus-specific amplification caused by repeated copies of a small DNA segment mapping within 9p12. Hum Gene. 2006;118:760-6.
18. Browne CE, Dennis NR, Maher E, Long SL, Nicholson J, Sillibourne J, et al. Inherited interstitial duplications of proximal 15q: genotype-phenotype correlations. Am J Hum Genet. 1997;61:1342-52.
]]>
19. Barber JCK, Joyce CA, Collinson MN, Nicholson JC, Willatt LR, Dyson HM, et al. Duplication of 8p23.1: a cytogenetic anomaly with no established clinical signiûcance. J Med Genet. 1998;35:491-6.
20. Barber JCK, Hall V, Maloney VK, Huang S, Roberts AM, Brady AF, et al. 16p11.2 - p12.2 duplication syndrome; a genomic condition differentiated from euchromatic variation of 16p11.2. Eur J Hum Genet. 2013;21:182-9.
21. Hysert M, Bruyere H, Cote GB, Dawson AJ, Dolling JA, Fetni R, et al. Prenatal cytogenetic assessment and inv(2)(p11.2q13). Prenat Diagn. 2006;26:810-3.
22. Entesarian M, Carlsson B, Mansouri MR, Stattin E-L, Holmberg E, Golovleva I, et al. A chromosome 10 variant with a 12 Mb inversion [inv(10)(q11.22q21.1)] identical by descent and frequent in the Swedish population. Am J Med Genet Part A. 2009;149A:380-6.
23. Djalali M, Steinbach P, Bullerdiek J, Holmes-Siedle M, Verschraegen-Spae MR, Smith A. The signiûcance of pericentric inversions of chromosome 2. Hum Genet. 1986;72:32-6.
24. Fickelscher I, Liehr T, Watts K, Bryant V, Barber JCK, Simone Heidemann, et al. The Variant inv(2)(p11.2q13) Is a Genuinely Recurrent Rearrangement but Displays Some Breakpoint Heterogeneity. Am J Hum Genet. 2007;81:847-56.
25. Gilling M, Dullinger JS, Gesk S, Metzke-Heidemann S, Siebert R,Meyer T, et al. Breakpoint cloning and haplotype analysis indicate a single origin of the common Inv(10)(p11.2q21.2) mutation among northern Europeans. Am J Hum Genet. 2006;78:878-83. ]]>
26. Mrasek K, Krüger G, Bauer I, Müller-Navia J, Liehr T, Weise A. A new unbalanced chromosomal abnormality in 1q31.1 to 1q32 without phenotypic consequences. Cytogenetic and Genome Research. 2008;121:286-7.
27. Daniel A, Darmanian A, Peters G, Goodwin L, Hort JR. An innocuous duplication of 11.2 Mb at 13q21 is gene poor: Sub-bands of gene paucity and pervasive CNV characterize the chromosome anomalies. Am J Med Genet Part A. 2007;143A:2452-9.
28. Roos A, Elbracht M, Baudis M, Senderek J, Schönherr N, Eggermann T, et al. A 10.7 Mb interstitial deletion of 13q21 without phenotypic effect defines a further non-pathogenic euchromatic variant. Am J Med Genet A. 2008;146A(18):2417-20.
]]>
29. Filges I, Röthlisberger B, Noppen C, Boesch N, Wenzel F, Necker J, et al. Familial 14.5 Mb interstitial deletion 13q21.1-13q21.33: clinical and array-CGH study of a benign phenotype in a three-generation family. Am J Med Genet A. 2009;149A(2):237-41.
30. Liehr T, Stumm M, Wegner RD, Bhatt S, Hickmann P, Patsalis PC, et al. 10p11.2 to 10q11.2 is a yet unreported region leading to unbalanced chromosomal abnormalities without phenotypic consequences. Cytogenet. Genome Res. 2009;124(1):102-5.
31. Liehr T, Bartels I, Zoll B, Ewers E, Mrasek K, Kosyakova N, et al. Is there a yet unreported unbalanced chromosomal abnormality without phenotypic consequences in proximal 4p? Cytogenet. Genome Res. 2011;132(1-2):121-3.
32. Barber JCK, Zhang S, Friend N, Collins AL, Maloney VK, Hastings R, et al. Duplications of proximal 16q flanked by heterochromatin are not euchromatic variants and show no evidence of heterochromatic position effect. Cytogenet Genome Res. 2006;114:351-8.
33. Rodríguez L, Niebuhr E, García A, Martínez-Fernández ML, Peña Segura JL. Be careful with familial unbalanced chromosome abnormalities!. Am J Med Genet Part A. 2008;146A:2005-7.
34. Warburton D. De novo balanced chromosome rearrangements and extra marker chromosomes identiûed at prenatal diagnosis: clinical signiûcance and distribution of breakpoints. Am J Hum Genet. 1991;49:995-1013.
]]>
Recibido: 20 de septiembre de 2013.
Aprobado: 3 de octubre de 2013.
Michel SorianoTorres. Instituto de Ciencias Básicas y Preclínicas "Victoria de Girón". Ave. 31 Esq. 146 No. 3102, Reparto Cubanacán, Playa, CP. 11400. La Habana, Cuba. Correo electrónico: michel.soriano@cngen.sld.cu ]]>