Fisiopatología de la drepanocitosis
Physiopathologic features of drepanocytemia
]]>
DraC. Eva Svarch
Instituto de Hematología e Inmunología. Ciudad de La Habana, Cuba.
]]>
RESUMEN
La hemoglobina (Hb) S es la consecuencia de una mutación puntual en la posición 6 de la cadena ß de la globina que resulta en la sustitución del ácido glutámico por la valina. La HbS polimeriza en condiciones de baja tensión de oxígeno y deforma al hematíe. El hematíe con Hb polimerizada tiene una sobrevida acortada y ocluye la microcirculación, lo que da lugar a la anemia hemolítica crónica y a las crisis vasooclusivas dolorosas que marcan en gran medida el cuadro clínico de la enfermedad. La oclusión vascular es un proceso complejo debido fundamentalmente a la prolongación del tiempo de tránsito de los hematíes a través de la microcirculación y/o a la disminución del tiempo de demora de polimerización de la HbS. En la actualidad se considera a la oclusión en la microcirculación como una forma de injuria de reperfusión en la que el estrés oxidativo y la inflamación llevan al daño crónico de los órganos. Los leucocitos y factores de la coagulación participan en el proceso. La anemia hemolítica contribuye a la oclusión vascular porque el aumento de reticulocitos, con más moléculas de adhesión en su superficie que las células maduras, determina su mayor adhesión al endotelio y porque la Hb libre en plasma consume óxido nítrico. El óxido nítrico actúa en diferentes formas, una de las más importantes es que desvía el balance normal entre vasodilatación-vasoconstricción hacia la vasoconstricción. Como consecuencia de estos hechos se describe un subfenotipo caracterizado por hipertensión pulmonar, accidente vascular encefálico, priapismo y úlcera maleolar. Existen factores genéticos que influyen en la severidad del cuadro clínico como la mutación 158 C-T en el gen de la globina g y la a talasemia. La primera aumenta la HbF y la segunda disminuye la CHCM, el número de células densas y la intensidad de la hemólisis. Sin embargo, la relación entre a talasemia y la severidad de la enfermedad no es clara.
Palabras clave: drepanocitosis, anemia hemolítica, oclusión vascular.
ABSTRACT
Hemoglobin S (Hb) is the consequence of a point mutation in of b globin chain 6 position resulting in glutamic acid substitution by valine. HB S polymerizes in conditions of oxygen low tension deforming the erythrocyte, the latter with the HB polymerized, has a shorten survival occluding the microcirculation, causing a chronic hemolytic anemia, and painful vasoconstrictive crises noting down to large extent the clinical picture of disease. Vascular occlusion is a complex process due mainly to transit time extension of erythrocytes through microcirculation and/or decrease of delayed time of polymerization of HbS. Nowadays, occlusion in the microcirculation is considered as an injury way of reperfusion in which oxidative stress of coagulation participates in the process. Hemolytic anemia contributes to vascular occlusion because of the reticulocytes, with more adhesive molecules in its surface than the mature ones, to determines its greater adhesion to endothelium, and because of the plasma free Hb to consume nitric oxide (NO). NO acts in different ways, where the most important is the deviation of normal balance between vasodilatation and vasoconstriction. As consequence from these facts we describe a sub-phenotype characterized by pulmonary hypertension, stroke, priapism, and malleolar ulcer. There are genetic factors influencing in clinical picture severity, e.g. 158 C-T mutation in globin gen g, and a-thalassemia. The first one increase the HBF, and the second one decrease the CHCM, the number of thick cells, and the hemolysis identity. However, relation between a-thalassemia and severity of disease is not clear.
]]>
Key words: Drepanocytemia, hemolytic anemia, vascular occlusion.
INTRODUCCIÓN
La hemoglobina (Hb) S es causada por una mutación puntual en el codon 6 del gen de la globina b que resulta en la sustitución de un solo nucleótido (GTG -- GTA) y como consecuencia, el remplazo del ácido glutámico por la valina en la superficie de la molécula. Los tetrámeros de hemoglobina se orientan de manera tal que en una de las 2 subunidades b, la valina, en la posición 6 forma un contacto hidrofóbico con un sitio complementario en la subunidad b del filamento adyacente.
La HbS desoxigenada polimeriza, esto deforma al hematíe que en determinadas circunstancias ocluye fundamentalmente la microcirculación y tiene una sobrevida acortada. Estos hechos dan lugar a las crisis vasooclusivas dolorosas (CVOD) y a la anemia hemolítica, que marcan en gran medida el cuadro clínico de la enfermedad.
Formación del polímero ]]>
La polimerización de la HbS es un proceso muy complejo que resulta en la formación de tetrámeros de HbS agregados o gelificados, en equilibrio con tetrámeros en solución. La transición de sol a gel de la HbS es la causa del aumento de la viscosidad de la sangre, de la distorsión del glóbulo rojo, del enlentecimiento del flujo circulatorio y de la oclusión vascular, con el consiguiente infarto de los órganos que caracterizan a la enfermedad.
Alteraciones en el nivel de oxígeno, pH, fuerza iónica, temperatura, 2, 3-difosfoglicerato, y concentración de HbS, afectan la formación del gel.1
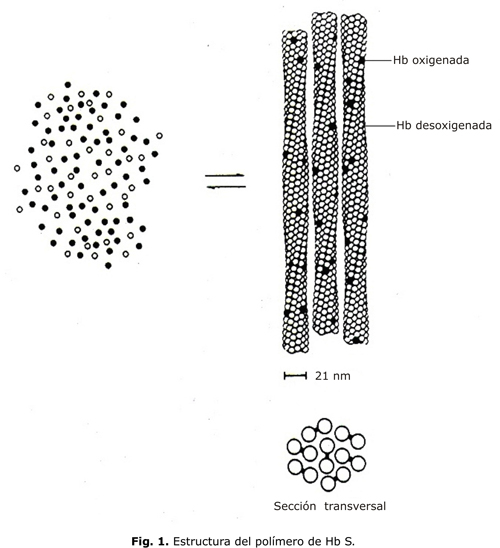
Con microscopía electrónica el polímero de HbS se ve como una estructura helicoidal formada por 7 pares de fibras que se orientan en el sentido longitudinal de la célula (fig. 1). Los contactos intermoleculares son de 3 tipos: a lo largo de un filamento, entre filamentos del mismo par y entre filamentos de pares diferentes.
Deformación del hematíe
La desoxigenación y consecuente polimerización de la HbS trae como consecuencia profundas alteraciones en la estructura y función de la membrana 2 y la deformación del hematíe que adopta diferentes formas según la velocidad en que la desoxigenación se produce. Si es muy rápida se formará el hematíe granular, si es menos rápida el hematíe en hojas de acebo (holly leaf) y si es lenta, el drepanocito reversible o irreversible (DI) (fig. 2). El DI es el que se observa en la lámina de sangre periférica, tiene un daño irreversible de la membrana, pero la Hb en su interior no está polimerizada. El número de DI se correlaciona con la magnitud de la hemólisis, pero no con la frecuencia de las CVOD.
 ]]>
]]>
Cinética de la polimerización
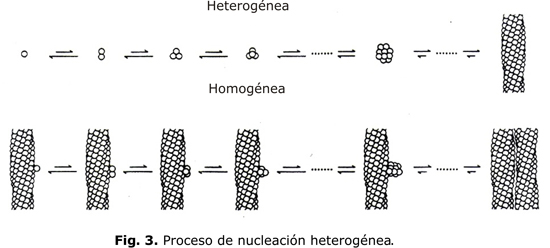
La cinética de la polimerización se puede explicar por el mecanismo de la doble nucleación. La polimerización se inicia por un proceso llamado de nucleación homogénea que tiene lugar en la HbS en solución y por el cual se agregan moléculas aisladas de HbS desoxigenada. Una agregación de pocas moléculas es termodinámicamente inestable, pero una vez que un cierto número de moléculas se agrega, se forma el núcleo crítico constituido por alrededor de 30 moléculas. El segundo proceso llamado de nucleación heterogénea tiene lugar en la superficie del polímero ya existente (fig. 3). Esta reacción es autocatalítica y produce un aumento exponencial del polímero. El resultado del mecanismo de la doble nucleación es un tiempo de demora entre el comienzo de la desoxigenación y la formación del polímero y su aumento exponencial con deformación de la célula. El tiempo de demora es de alrededor de 30 seg. Pequeños cambios en la concentración de Hb tienen un marcado efecto en el tiempo de demora; por ejemplo si la CHCM disminuye de 32g/dL a 30g/dL, el tiempo de demora aumenta 3 veces.3 El fenómeno de la polimerización retardada de la HbS en solución se observa también dentro de los hematíes. Después de una desoxigenación rápida, el tiempo en que los hematíes se deforman es de alrededor de 30 seg.4 La cinética de esta reacción desempeña un papel crítico en la reología y morfología de los hematíes circulantes y por lo tanto, en la fisiopatología de la oclusión vascular.5
La oxigenación y desoxigenación de los glóbulos rojos circulantes se produce más o menos en el mismo tiempo en que tiene lugar la falciformación y reversión a la normalidad in vitro. Los drepanocitos expuestos a altas tensiones de oxígeno en los pulmones vuelven a la forma normal aproximadamente en 0,5 seg y se mantienen como discocitos mientras se encuentran a la presión de oxígeno de la circulación arterial. Cuando entran en los capilares, la saturación de oxígeno disminuye rápidamente y disminuye la solubilidad de la Hb S. Los glóbulos rojos demoran aproximadamente un segundo en atravesar la microcirculación, aunque este tiempo es muy variable. Dado que el tiempo de demora en condiciones basales es de alrededor de 30 seg, la mayoría de los hematíes la atraviesan indemnes.
Es importante tener en cuenta que existe polímero en discocitos a saturaciones de oxígeno más altas que las requeridas para la deformación del hematíe.
No parece existir una relación muy clara entre la fracción de HbS polimerizada en el glóbulo rojo y la severidad clínica.6 ]]>
Si por alguna razón se prolonga el tiempo de tránsito o disminuye el tiempo de demora, todos los glóbulos rojos tendrán Hb polimerizada en su interior, se deformarán y ocluirán la microcirculación. Si el tiempo de tránsito por la circulación capilar es corto, no se producirá oclusión. Esto es probablemente lo que ocurre en el miocardio. El infarto de miocardio es raro en la drepanocitosis, a pesar de que en el miocardio, la PO2 es baja. Esto se debe a que el tiempo de tránsito por la circulación es muy corto.En el entorno de la microcirculación, la viscosidad interna del glóbulo rojo y la elasticidad de la membrana son más importantes que la viscosidad de la sangre total.
Interacción de la HbS con la HbA y la HbF
La HbA y la HbF tienen un efecto importante sobre el tiempo de demora que depende de su concentración dentro del hematíe. El efecto de los tetrámeros de HbF y de sus híbridos asimétricos a2 bs bg es mucho mayor que el de la HbA. En determinadas condiciones, la HbA puede formar parte del polímero de HbS, pero la HbF es siempre excluída. Por esta razón, la S persistencia hereditaria de la HbF (SPHHbF) es prácticamente asintomática. En la actualidad es posible aumentar farmacológicamente el nivel de HbF y por lo tanto, aumentar el tiempo de demora y disminuir la polimerización de la HbS. Es probable que en el futuro se pueda correlacionar el tiempo de demora y la fracción de polímero dentro del hematíe con la severidad clínica de la enfermedad.1
]]>
La oclusión vascular es un proceso complejo en el que participan muchos factores: la deshidratación del glóbulo rojo, alteraciones de su membrana, aumento de su adhesión al endotelio, alteraciones intrínsecas de las células endoteliales y otros.
Homeostasis anormal de cationes
Una homeostasis anormal de cationes lleva a la deshidratación de los hematíes con formación de células densas en general y drepanocitos irreversibles en particular. Estas células influyen en la prolongación del tiempo de tránsito de la sangre a través de la microcirculación y también en el grado de hemólisis. ]]>
Los mecanismos que contribuyen a la deshidratación del GR son el cotransporte de Cl-K y la salida de K activada por el Ca++ (cadena Gardos).7En el estado basal del paciente el Ca++ está aumentado en los GR SS, pero dentro de vesículas intracelulares con concentraciones normales en el citoplasma. Cuando la membrana se deforma, hay un aumento transitorio del Ca++ en el citoplasma. Este aumento es suficiente para activar la cadena Gardos que induce la salida de K y agua de la célula y por consiguiente, la deshidratación, falciformación y formación de células densas.7 La activación combinada del cotransporte de Cl-K y de la cadena Gardos lleva a la rápida deshidratación de una subpoblación de drepanocitos jóvenes que se transforman en drepanocitos irreversibles.8
El cotransporte de Cl-K es activado por edema de la célula y acidosis, y la cadena Gardos por moléculas proinflamatorias, lo que explica la asociación entre infección, CVOD y aumento de la hemólisis.9
El cotransporte de Cl-K es inhibido por el magnesio y la cadena Gardos por un antifúngico: el cotrimazol.
En la figura 4 se muestran los principales mecanismos que intervienen en la deshidratación del glóbulo rojo.
Difunción de la bicapa lipídica
En el estado basal los fosfolípidos que contienen colina (esfingomielina y fosfatidilcolina), están localizados en la capa externa de la membrana, mientras los que contienen aminofosfolípidos (fosfatidilserina y fosfatidiletanolamida), están en la capa interna. En la drepanocitosis hay una pérdida de la asimetría de la membrana con exposición de la fosfatidilserina en la superficie, lo que aumenta la adhesión de los glóbulos rojos entre sí y a las células endoteliales, y condiciona un fenotipo procoagulante del eritrocito.10 ]]>
Esta estructura de la membrana se mantiene por un sistema de transporte activo que mueve los fosfolípidos a través de la membrana; la flipasa que fija la fosfatidilserina en la superficie externa de la membrana y la escramblasa, que mueve los fosfolípidos de la capa interna a la externa y viceversa.11 La HbF protege contra el movimiento de la fosfatidilserina hacia la superficie de la membrana.Esta bicapa lipídica disfuncional contribuye en gran medida a la fisiopatología de la enfermedad.
Otras alteraciones de la membrana
La HbS polimerizada se une a la capa interna y es el factor más importante que determina la rigidez de la membrana.
La HbS es inestable y tiene tendencia a la desnaturalización formando pequeños cuerpos de Heinz que se unen con agregados de banda 3. Los agregados de banda 3 en la superficie de la célula se unen con anticuerpos anti-banda 3, un proceso que también ocurre en el envejecimiento de las células normales y que contribuye a la sobrevida acortada de los glóbulos rojos SS. Los anticuerpos anti-banda 3 disminuyen la adhesión de los hematíes al CD36 de las células endoteliales.12 Por esta razón, los agregados de banda 3 pueden ser un factor importante en el desencadenamiento de las CVOD, pero pueden intervenir también en su resolución.
Los cuerpos de Heinz secuestran lípidos, espectrina, ankirina y proteína 4.1. ]]>
La HbS tiene tendencia a autooxidarse, forma metahemoglobina y se generan potentes oxidantes como superóxido, peróxido y radicales hidroxilo, que producen un daño adicional de la membrana.2
Reacciones de adhesión
Desde hace muchos años se conoce que los glóbulos rojos SS se adhieren al endotelio in vitro, y que esta adhesión se correlaciona con la severidad de la enfermedad.13 Las interacciones adhesivas son complejas e involucran a una gran cantidad de ligandos, receptores e interacciones no específicas que varían dependiendo de la edad y densidad de la célula, las alteraciones del endotelio, factores en el plasma y la velocidad del flujo circulatorio.1 La adhesión ocurre entre los hematíes y las células endoteliales entre los hematíes y la matriz subendotelial que se expone por injuria de las células endoteliales o por retracción inducida por trombina, y en determinadas condiciones, entre los hematíes entre sí.
Los reticulocitos y glóbulos rojos jóvenes son los que se adhieren primero porque tienen más receptores que involucran moléculas de adhesión en su superficie; estos disminuyen a medida que la célula envejece y deviene más densa.14 Posteriormente, la adhesión de agregados heterocelulares de células densas, drepanocitos irreversibles y leucocitos, aumenta la oclusión vascular, lo que da como resultado hipoxia local, acidosis, aumento de la polimerización de la HbS y propagación de la oclusión a los vasos adyacentes.15
Las moléculas más importantes que promueven la adhesión en el hematíe son:
- CD 47 ]]>
- CD36- a4 b1
- Glicolípidos sulfatados
- Fosfatidilserina
- Agregados de banda 3
- BCAM/Lu
El BCAM/Lu (basal cell adhesion molecule) es portador del grupo sanguíneo Lutheran.
La integrina a4 b1 se une a su ligando en el endotelio, el VCAM-1.16 La p-selectina participa en la interacción entre glóbulos rojos, leucocitos, plaquetas y células endoteliales. Los glicolípidos sulfatados y la fosfatidilserina son proadhesivos. ]]>
Las moléculas más importantes que promueven la adhesión en las células endoteliales son las siguientes:- VCAM-1
- CD36
- aV b3
- P-selectina
- Laminina
- Fibronectina
El VCAM-1 no se expresa constitutivamente, pero lo hace por la acción de citocinas anti-inflamatorias como el FNT a y la IL8 y por hipoxia. La hipoxia aumenta la adhesión del VCAM-1 a la molécula de adhesión a4 b1 del glóbulo rojo.17 La fibronectina y la laminina se encuentran en la matriz subendotelial y se unen con el hematíe a través del BCAM-1.
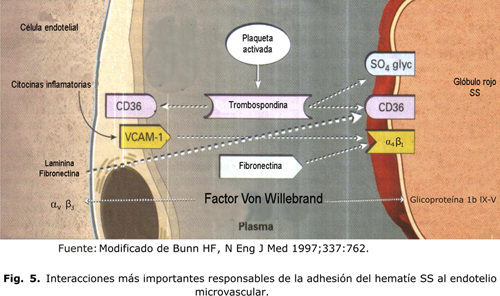
Las moléculas de unión en el plasma son la trombospondina, la fibronectina, la laminina solubles y el factor Von Willebrand. Un mecanismo bien identificado es el papel de unión de la trombospondina soluble entre el CD36 del glóbulo rojo y varios receptores endoteliales que se expresan constitutivamente incluyendo la vitronectina (av b3) y el CD36. Los multímeros de alto peso molecular del factor Von Willebrand promueven también la adhesión del hematíe a la molécula de adhesión av b3.18 En la figura 5 se muestran las interacciones más importantes responsables de la adhesión del hematíe SS al endotelio microvascular. ]]>
El proceso de adhesión puede dislocar las células endoteliales y que estas aparezcan circulando en sangre periférica con un fenotipo activado que expresa las moléculas de adhesión ICAM-1, VCAM-1, E-selectina, P-selectina y factor tisular.19
La epinefrina modula los procesos adhesivos, lo que explica por qué el estrés puede desencadenar CVOD.
Alteraciones del endotelio
El endotelio participa activamente en el proceso de la oclusión vascular, presenta alteraciones histológicas en casi todos los órganos y en los sitios en que existe hiperplasia de la íntima se observa la formación de trombos.20 ]]>
Además, el endotelio se encuentra bajo la influencia de un conjunto de estímulos que lo conducen a un estado de activación crónica, con un número alto de células endoteliales que expresan las moléculas de adhesión VCAM-1, ICAM-1, E selectina y P selectina.En la tabla 1 se muestran las causas de activación del endotelio.
La activación del endotelio tiene lugar a través de un factor de transcripción "nuclear factor kappa B" (NFkB), del factor de crecimiento rápido del endotelio (EGR-1) y del factor de activación de la proteína (AP-1). El endotelio activado tiene un fenotipo proinflamatorio, proadhesivo y procoagulante que provee el marco disfuncional en el que la oclusión vascular tiene lugar.
Estímulos inflamatorios y citocinas como el FNTa, las IL1 y la IL8 activan las células endoteliales. Esto explica por qué son frecuentes las CVOD durante las infecciones bacterianas.
La disminución del óxido nítrico y el aumento de la endotelina-1 desempeñan un papel esencial en el tono vasomotor y en las alteraciones del endotelio. El óxido nítrico es un potente vasodilatador y regula la adherencia de las células, la agregación plaquetaria y la producción de eicosanoides como la prostaciclina.9 La endotelina-1 es un potente vasoconstrictor y es proinflamatoria. La hidroxiurea disminuye la concentración de endotelina-1.
Las causas más importantes de adhesión al endotelio se muestran en la tabla 1.
Modelos experimentales de hipoxia reoxigenación apoyan la hipótesis de que la oclusión microvascular es una forma de injuria de reperfusión en la que el estrés oxidativo y la inflamación llevan al daño crónico de los órganos.21
]]> Leucocitos
La leucocitosis se correlaciona de manera independiente con la severidad del cuadro clínico, con la mortalidad, con la CVOD y con el síndrome torácico agudo. Después de la administración de G-CSF se ha observado síndrome torácico agudo y fallo multiorgánico. Los leucocitos presentan un fenotipo activado: degranulación, alteraciones en la regulación de la L selectina, una glicoproteína que inicia la adhesión de los leucocitos al endotelio y un aumento del leucotrieno B4.22
Alteraciones de la coagulación
Aunque los estudios de las anormalidades de la coagulación no son concluyentes, es muy posible que, sobre todo durante las CVOD, los factores procoagulantes, anticoagulantes y las plaquetas desempeñen un papel en la oclusión vascular. Esto es particularmente importante en las interacciones que tienen lugar en la superficie del endotelio, donde el factor Von Willebrand, las plaquetas y la trombospondina aumentan la adhesividad.
La oclusión vascular es favorecida por la generación de trombina in vivo.23 El endotelio activado y la fosfatidilserina en la superficie de los hematíes condicionan un estado trombofílico.24
La heparina inhibe la adhesión entre los glóbulos rojos y el endotelio in vitro a través de la P selectina.25 En un estudio preliminar, el aumento del fragmento 1.2 de la protrombina y el aumento de la fosfatidilserina del glóbulo rojo se correlacionan con el aumento de la velocidad del flujo sanguíneo medido por ultrasonido doppler transcraneal en algunas arterias cerebrales.26 ]]>
Hay evidencias de que el factor activador de las plaquetas aumenta la adherencia de los hematíes al endotelio y que esta actividad puede ser bloqueada selectivamente por un anticuerpo anti- integrina av b3 lo que convertiría a este anticuerpo en una probable arma terapéutica.14Un resumen de los distintos factores que condicionan la oclusión vascular se expone en la tabla 2.
Anemia hemolítica
Como ya se mencionó, la anemia hemolítica se debe a profundas alteraciones de la membrana del glóbulo rojo y es fundamentalmente extravascular por eritrofagocitosis, pero también intravascular.
Durante mucho tiempo se consideró que la anemia hemolítica desempeñaba un papel secundario en el cuadro clínico de la drepanocitosis en relación con los fenómenos vasooclusivos. Nuevos conocimientos sobre la hemólisis y sus efectos en la biología del óxido nítrico y un examen más profundo de los subfenotipos de la enfermedad requiere una nueva evaluación del cuadro clínico.
El papel que desempeñan una homeostasis anormal del óxido nítrico y la adherencia de los reticulocitos en la fisiopatología de la drepanocitosis, han relacionado directamente la magnitud de la hemólisis con los fenómenos vasooclusivos. Normalmente el óxido nítrico se une con la guanilato ciclasa soluble que transforma la guanosina-trifosfato en guanosina monofosfato cíclica. Estas enzimas, que participan en el metabolismo de los nucleótidos, producen relajación del músculo liso y vasodilatación. Cuando la Hb plasmática liberada por la hemólisis intravascular consume óxido nítrico, el balance normal entre vasoconstricción y vasodilatación es desviado hacia la vasoconstricción. De esta manera, la anemia hemolítica y la oclusión vascular se sobreponen en la propensión a padecer los subfenotipos de hipertensión pulmonar, accidente vascular encefálico, priapismo y úlcera maleolar. La a talasemia, que disminuye la hemólisis y mejora la anemia, disminuye la incidencia de las 3 últimas complicaciones, mientras que la HbF desempeña un papel menos prominente. Otras manifestaciones asociadas con un aumento de la viscosidad sanguínea como las CVOD, el síndrome torácico agudo y la osteonecrosis, no se relacionan con la magnitud de la hemólisis. En estas manifestaciones el aumento de HbF disminuye su frecuencia y severidad.26
Acciones terapéuticas que hagan menos intensa la hemólisis o aumenten la biodisponibilidad del óxido nítrico, pueden disminuir la incidencia o severidad de los subfenotipos relacionados con la hemólisis en la drepanocitosis.27 ]]>
Factores genéticos que influyen en la variabilidad del cuadro clínico
La producción de HbS es un evento monogénico, es indispensable, pero insuficiente para determinar el fenotipo.28 Otros genes no ligados al locus de la globina b llamados pleiotrópicos o efectores secundarios, intervienen en diferentes procesos como la velocidad de destrucción de los hematíes, la formación de células densas, la adherencia al endotelio vascular y otros involucrados en la fisiopatología. No todos los pacientes tienen los mismos genes pleiotrópicos, por lo que la severidad de la AD es muy variable. En el estudio del perfil genético se pueden identificar estos genes y también los epistáticos o modificadores que ayudan a definir el riesgo individual.9 Ejemplo de genes epistáticos son la mutación 158 C-T en el gen g, que determina un aumento de la HbF sobre todo en pacientes con el haplotipo Senegal y asiático, y la a talasemia, que disminuye la CHCM, el número de células densas y la intensidad de la hemólisis. Sin embargo, a pesar del efecto beneficioso de la a talasemia en muchos aspectos que intervienen en la oclusión vascular, su relación con la severidad del cuadro clínico no es clara y esto se debe a la complejidad de la fisiopatología de la enfermedad.
REFERENCIAS BIBLIOGRÁFICAS
1. Nathan DG, Orkin SH, Ginsburg D, Look AT. Hematology of infancy and childhood. Saunders. Philadelphia 2003. pp. 792-826.
]]>
2. Hebbel RP. Beyond hemoglobin polymerization: The red blood cell membrane and sickle disease pathophysiology. Blood 1991;77:214-37.
3. Eaton WA, Hofrichter J. Hemoglobin S gelation and sickle cell disease. Blood 1987;70:1245-66.
4. Rampling MW, Sirs JA. The rate of sickling of cells containing sickle-cell hemoglobin. Clin Sci Mol Med 1973;45:655-64.
5. Eaton WA, Hofrichter J. Sickle cell hemoglobin polymerization. Adv Protein Chem 1990;40:63-279.
6. Poillon WN, Kim BC, Castro O. Intracellular hemoglobin S polymerization and the clinical severity of sickle cell anemia. Blood 1998;91:1777-83.
]]>
7. Bunn HF. Pathogenesis and treatment of sickle cell disease. N Eng J Med 1997;337:762:9.
8. Bookchin RM, Lew VL. Sickle red cell dehydration: Mechanisms and interventions. Curr Opin Hematol 2002;9:107-10.
9. Stuart MJ, Nagel RL. Sickle cell disease. Lancet 2004;364:1343-60.
10. Setty BNY, Kulkam S, Stuart MJ. Role of erythrocyte phosphatidylserine in sickle red cell endothelial adhesión. Blood 2002;99:1564-71.
11. De Jong K, Larkin SK, Styles FA, Bookchin RM, KuypersFA. Characterization of the phosphatidylserine-exposing subpopulation of sickle cells. Blood 2001;98:860-7.
]]>
12. Hornig R, Lutz HU. Band 3 protein clustering on human erythrocytes promotes binding of naturally occurring anti-band 3 and anti-spectrin antibodies. Exp Gerontol 2000;35:1025-44.
13. Hebbel RP, Boogaerts MA, Eaton JW, Steinberg MH. Erythrocyte adherence to endothelium in sickle cell anemia: A possible determinant of disease severity. N Eng J Med 1980;302:992-5.
14. Kaul DK, Tsai HM, Liu XD, Nakada MT, Nagel RL, Coller BS. Monoclonal antibodies to avb3 (7E 3 and LM 609) inhibit sickle red blood cell-endothelial interaction induced by platelet-activating factor. Blood 2000;95:368-74.
15. Turban A, Weiss LA, Mohandas N, Coller BS, Frenette PS. Primary role of adherent leucocytes in sickle cell vascular occlusion: A new paradigm. Proc Natl Acad Sci USA 2002;99:3047-51.
16. Gee BE, Platt OS. Sickle reticulocytes adhere to VCAM-1. Blood 1995;85:268-74.
]]>
17. Setty BN, Stuart MJ. Vascular cell adhesion molecule-1 is involved in mediating hypoxia-induced sickle red blood cell adherence to endothelium: Potential role in sickle cell disease. Blood 1996;88:2311-20.
18. Wick TM, Moake JL, Udden MM, Mc Intire IV. Unussually large Von Willebrand factor multimers preferentially promote young sickle and non-sickle erythrocyte adhesión to endothelial cells. Am J Hematol 1993;42:284-92.
19. Solovey A, Lin Y, Browne P, Choong S, Wainer E, Hebbel RP. Circulating activated endothelial cells in sickle cell anemia. N Eng J Med 1997;337:1584-90.
20. Rothman SM, Fulling KH, Nelson JS. Sickle cell anemia and central nervous system infarction: A neuropathological study. Ann Neurol 1986;20:684-90.
21. Kaul DK, Hebbel RP. Hipoxia/reoxigenation causes inflamatory response in transgenic sickle mice but not in normal mice. J Clin Invest 2000;106:411-20.
]]>
22. Setty BNY, Stuart MJ. Eicosanoids in sickle cell disease: Potencial relevance of neutrophil leukotriene B, to disease pathophysiology. J Lab Clin Med 2002;139:80-9.
23. Tomer A, Harker LA, Eckman JR. Thrombogenesis in sickle cell disease. J Lab Clin Med 2001;137:398-407.
24. Matsui NM, Varki A, Embury SH. Heparin inhibits the flow adhesión of sickle red blood cell to P-selectin. Blood 2002;100:3790-6.
25. Styles L, de Jong K, Vishinky E. Increased RBC phosphatidylserine exposure in sickle cell disease patients at risk for stroke by transcranial Doppler screening. Blood 1997;90:604(abstr).
26. Kato GJ, Gladwin MT, Steinberg MH. Deconstructing sickle cell disease: Reappraisal of the role of hemolisis in the development of clinical subphenotypes. Blood Rev 2007;21:37-47.
]]>
27. Lapoomérouli C, Benkrrow M, Odievre MH, Ducrocq R, Brun M, Elion J. Decreased plasma endothelin-1 levels in children with sickle cell disease treated with hydroxyurea. Haematologica 2005;90:401-3.
28. Chui DH, Dover GJ. Sickle cell disease: No longer a single gene disorder. Curr Opin Pediatr 2001;13:22-7.
]]>
Recibido: 12 de enero del 2009.
DraC. Eva Svarch. Instituto de Hematología e Inmunología. Apartado Postal 8070, Ciudad de La Habana, CP 10800, Cuba. Tel (537) 6438268, 6438695. Fax (537) 6442334. e-mail: ihidir@hemato.sld.cu. Sitio Web: http://www.sld.cu/sitios/ihi ]]>